

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
半三明治配合物的芳香性及其与环伸缩振动拉曼光谱频率的相关性
[韩立楠 , 刘子忠
, 刘子忠* , 刘红霞, 沈臣飞]
, 刘子忠, 刘红霞, 沈臣飞]
|
|


作者简介: 韩立楠, 1992年生, 内蒙古师范大学化学与环境科学学院硕士研究生 e-mail: hln503586588@163.com
寻找实验上定量测定芳香性分子芳香性大小的方法是科学家一直追求的一个目标, 应用拉曼光谱测量半三明治配合物芳香性大小是一项新的尝试。 采用Gaussian09计算程序中的密度泛函理论(DFT), 对(η6-C6X6)和半三明治配合物[(η6-C6X6)M] n+(X=F~Br, M=Ti~Mn, n=1, 2)进行几何优化, 并对其几何结构、 静电引力、 稳定化能Δ E、 核独立化学位移(NICS)值及环伸缩振动拉曼光谱频率(RSVRSF)大小进行了理论计算。 结果表明: 取代苯及其形成的半三明治配合物的NICS值均为负值, 均为芳香性分子; 取代苯及其半三明治配合物中均存在 A1g或 A1对称性的RSVRSF, 且其峰强度均很大, 其NICS绝对值、 RSVRSF值均随着F, Cl, Br取代的顺序逐渐减小, 均呈高度正相关, 且相关性系数均达到0.99以上, 理论预测可通过实验测定其RSVRSF值测定该类物质的芳香性大小。
It is a pursuing goal to find an experimental method for determining the aromatic size of aromatic molecules for scientists. It is a new attempt to measure the aromaticity degree of half-sandwich complexes using Raman spectroscopy.The calculations for the geometries optimization, electrostatic forces, stabilization energies, nucleus independent chemical shifts (NICS) and values of the Raman spectroscopy for (η6-C6X6) and half sandwich complexes [(η6-C6X6)M] n+(X=F~Br, M=Ti~Mn, n=1, 2) were conducted using density functional theory (DFT) method within Gaussian09 process package. The results showed that, for all the (η6-C6X6) and half sandwich complexes, the values of NICS were all negative. These kinds of molecules all were aromatic. There were strong Ring Stretching Vibration Raman Spectroscopic Frequency (RSVRSF) peaks with A1g/ A1 symmetric in the (η6-C6X6) and its half sandwich complexes. The RSVRSF values and the absolute NICS values of the (η6-C6X6) and its half sandwich complexes gradually decreased with the order of F, Cl, and Br, showing a highly positive correlation, and the correlation coefficients were above 0.99. It is theoretically predicted that the determination of the aromatic degree is possible by the experiment determination of the RSVRSF values for the (η6-C6X6) and its half sandwich complexes.
自从1919年Hein等分离出第一个芳香性金属化合物, 引发了科学研究芳香性金属化合物(三明治、 半三明治配合物)的新热潮, 一直以来这类物质的研究就从未被忽视, 尤其是近几年关于半三明治配合物的合成、 计算研究越来越多, 发现半三明治配合物在药物、 材料、 纳米化学等研究领域中都有着极为重要的研究意义[1]。 如Horvá thová 等应用密度泛函理论不同计算方法计算研究了中性和带正电的V-benzene和Co-benzene半三明治配合物的结构、 能量等性质, 从能量角度推断出几何结构相似的V-benzene和Co-benzene体系均存在多种不同的电子结构[2], 为实验合成不同种过渡金属-苯类型的半三明治配合物提供了较为扎实的理论基础; 孙章等从实验上应用激光烧灼法合成了M[C6F6]-(M=Ag, Au)半三明治配合物, 又应用密度泛函理论计算了其结构, 发现分子的光电子能谱(PES)、 绝热电子亲合势(EAS)的实验测量值与理论计算值均一致, 不仅合成了新的半三明治配合物为后续取代苯形成半三明治配合物提供实验方法, 还验证了理论研究该类物质方法的可靠性[3], 越来越多的科研工作者将理论与实验相结合, 研究分子的振动光谱、 结构等性质, 计算为实验提供理论基础, 实验验证计算的可靠性; 2011年张敏[4]、 刘玉宁[5]等理论研究了以六氟代苯和(η 6-1, 3, 5-C3P3H3)为配体与V、 Cr形成的半三明治、 三明治配合物的结构、 键能、 芳香性等性质, 发现配合物均为芳香性分子, 并且随着F取代数目的增加芳香性增大; 其中用来判断半三明治配合物芳香性的依据是核独立化学位移(NICS)[6, 7, 8, 9, 10, 11], 它是一种从理论上预测分子芳香性的判据, 但无法从实验上测量物质的NICS值。 到目前为止还没有一种十分准确的方法能够在实验上定量测量半三明治配合物芳香性大小的方法, 而且以(η 6-C6Cl6), (η 6-C6Br6)为配体的半三明治配合物的结构、 芳香性、 光谱等性质仍然没有得到系统的研究。
基于以上问题, 本研究应用量子化学方法, 研究[(η 6-C6X6)M]n+(X=H, F~Br, M=Ti~Mn, n=1, 2)半三明治配合物的结构、 芳香性、 拉曼光谱等性质, 以期望从理论上寻找到一种能够在实验上定量测量半三明治配合物芳香性大小的新方法。
应用Gaussian09量子化学计算程序, 密度泛函理论(DFT)B3LYP计算方法, 有H原子的体系应用6-311++G(d, p)基组, 无H原子体系应用6-311+G(d)基组对目标分子进行几何结构优化以及A1g/A1对称类型的环伸缩振动拉曼光谱频率(RSVRSF)计算; 几何结构优化到最高对称性的基础之上采用B3LYP-GIAO方法计算目标分子的核独立化学位移(NICS), 同时应用Gaussian09量子化学计算程序中的 NBO3.1程序, B3LYP计算方法计算目标分子的电荷、 轨道系数、 成键情况等; 应用GaussView5.0绘制目标分子几何结构图、 特征分子轨道图; 采用社会科学软件包SPSS17.0计算目标分子的NICS与其RSVRSF之间的相关系数; 最后用Origin8.0绘制相关性曲线。
2.1.1 几何结构
所有配体均属于D6h点群, 几何结构见图1。 没有虚频, 能稳定存在。 取代苯的C— C和C— X键长均随着F, Cl和Br取代的顺序逐渐增大, 分析键长变化原因, 键长即两原子间核间距, C— C键长变化微小, 主要是因为C— C键的成键原子均为C原子, 核间距变化微小, 而C— X键的成键原子电子层数随着F, Cl和Br顺序逐渐增大导致C— X键核间距增大, 即键长逐渐增大, H原子的电子层数少, C— H键之间的核间距较短, 导致C— H键长较短(见图1)。
 | 图1 (η 6-C6X6), [(η 6-C6X6)Ti]2+优化几何结构、环伸缩振动模式Fig.1 Optimized geometries and ring stretching vibration mode for (η 6-C6X6), [(η 6-C6X6)Ti]2+ |
2.1.2 电子结构
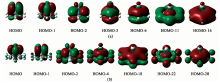
图2是配体的部分特征分子轨道图, 均有6个π 电子满足休克尔(4n+2)π 电子规则, 其中(η 6-C6H6)的HOMO-4, (η 6-C6F6)的HOMO-2这2个π 分子轨道均是由C原子的pz轨道肩并肩形成的共轭大π 键, 诱导产生π 电子环流, 表现出π 芳香性。 与苯的HOMO-4相比较(η 6-C6F6)的HOMO-14不仅有C原子的pz轨道, 还有F原子的pz轨道共同参与形成, 诱导产生更大的π 电子环流, 表现出更强的π 芳香性; 其中(η 6-C6H6)的HOMO-14和C6F6的HOMO-26是σ 分子轨道, 诱导产生σ 环流, 表现出σ 芳香性, 苯的H原子参与形成了σ 分子轨道, 而F原子没有参与形成σ 分子轨道; 氯苯、 溴苯的π 芳香性与氟苯相似, σ 芳香性与苯相似。
 | 图2 (η 6-C6X6)的特征分子轨道图 (a): (η 6-C6H6); (b): (η 6-C6F6)Fig.2 Characteristic molecular orbitals for (η 6-C6X6) (a): (η 6-C6H6); (b): (η 6-C6F6) |
为了进一步探讨配体芳香性, 分析其自然键轨道(natural band orbital, NBO)电荷转移情况, 以苯、 氟苯中原子NBO电荷的自然排布为例:
(η 6-C6H6):
C [core]2s(0.97)2p(3.25[2
H 1s(0.76)
(η 6-C6F6):
C [core]2s(0.84)2p(2.82)[2
F [core]2s(1.83)2p(5.47)[2
(η 6-C6H6)中C原子的2s轨道中电子数目相比较于未成键的自由C原子的2s轨道中电子数目减小1.03e, H原子的1s轨道中电子数目减小0.24e, 共减小1.27e, 而C的2p轨道中的电子数目增加了1.25e, 其中2pz电子数目不变, 2px和2py共增加了1.25e, 即(η 6-C6H6)中C和H原子的s轨道中电子主要转移到了C的2px和2py轨道中, 展现了苯的σ 芳香性, 如(η 6-C6H6)的HOMO-14, 苯中C原子的px和py轨道参与形成诱导产生的σ 环流, 其余减小部分转移到了C的极化函数3d和4p轨道中; 同理(η 6-C6F6)中C和F原子的2s轨道中电子、 F的2px和2py轨道中电子主要转移到了其C的2px和2py以及F的2pz轨道中, 既说明了氟苯中C的2px和2py轨道参与形成了σ 分子轨道, 诱导产生σ 环流, 与苯相似存在σ 芳香性, 又说明了氟苯中F原子的2pz轨道也参与了C原子2pz轨道之间形成的共轭大π 键, 诱导产生了更大的π 电子环流, 如(η 6-C6F6)的HOMO-14, 而F原子的2px和2py共减少0.45e转移到其2pz轨道中, 并且没有参加形成σ 分子轨道如(η 6-C6F6)的HOMO-26, 即与苯相比较氟苯中F原子的2pz参与了其环上碳碳共轭的π 电子环流的形成, 增加了其π 芳香性, 而对其σ 芳香性影响不大, 其余减小部分转移到了C的极化函数3d和4p轨道中; 氯苯、 溴苯中电子转移情况与苯、 氟苯相似。
配体分子中C, H和X的s轨道中减少的电子主要转移到其p轨道中, 其p轨道中电子数目增加由多到少的顺序是: (η 6-C6F6)> (η 6-C6Cl6)> (η 6-C6Br6), 而芳香性受C, X的p轨道参与成键情况以及其中电子数目影响, 其p轨道中电子数目增加的顺序与其芳香性大小密切相关, 进一步研究发现其p轨道中电子数目增加由多到少的顺序与其芳香性顺序一致(η 6-C6F6)> (η 6-C6Cl6)> (η 6-C6Br6)。
2.2.1 几何结构
半三明治配合物均属于C6v点群, 几何结构见图1。 其中[(η 6-C6Br6)Ti]2+有一个虚频为过渡态, [(η 6-C6X6)Cr]+系列半三明治配合物均存在两个虚频(NIF)见表2, 其余分子均没有虚频(见图1)。 与配体相比较, C— C键长均有所增大, C— H键长基本不变, C— X键长均有所减小, 见表1和表2。 金属离子的加入使其C— C键长增大, 作用力减小, 如(η 6-C6H6)和(η 6-C6F6)的WBIC-C分别为1.438和1.350, [(η 6-C6H6)Ti]2+, [(η 6-C6F6)Ti]2+的WBIC— C分别为1.345和1.250, 分别减小了约0.1, 即金属离子的加入活化了其C— C键。 分析变化原因, 过渡金属离子与配体之间不仅存在静电引力见表2, 还存在一定的共价作用见图3, 即与配体相比较, 不仅C原子之间成键、 受取代原子变化影响, C原子还与金属离子成键, 此时C原子形成C— C键的成分就会有所减小, 所以C— C键的强度、 作用力就会减弱, C— C键的键长就会增大, 以氯苯、 溴苯为配体形成的半三明治配合物的键长变化规律与氟苯相似不做一一解释。
| 表1 (η 6-C6X6)的点群、 虚频(NIF)、 键长(Å )、 理论计算NICS和(A1g)RSVRSF Table 1 Point group, the number of imaginary frequencies (NIF), bond lengths (Å ), theoretical values of the RSVRSF (A1g) (cm-1), force constants and NICS (ppm) for (η 6-C6X6) |
| 表2 [(η 6-C6X6)M]n+的虚频、 键长(Å )、 WBI、 静电引力V、 稳定化能Δ E (kJ· mol-1)、理论计算RSVRSF(A1)(cm-1)和NICS(ppm) Table 2 NIF, bond lengths (Å ), WBI, electrostatic forces stabilization energies Δ E (kJ· mol-1), the theoretical values of the RSVRSF(A1)(cm-1), force constants and NICS(ppm) for [(η 6-C6X6)M]n+ |
相同配体不同过渡金属离子形成的半三明治配合物的C— C, C— X键长变化无明显变化规律, 而不同配体相同过渡金属离子形成的半三明治配合物C— C, C— X键长变化有一定规律, 其RC— C, RC— X键长由大到小的顺序均是: [(η 6-C6Br6)M]n+> [(η 6-C6Cl6)M]n+> [(η 6-C6F6)M]n+, [(η 6-C6H6)M]n+的RC— C, RC— X键长小于[(η 6-C6F6)M]n+的RC— C, RC— X键长, 即取代苯形成的半三明治配合物C— C键活化程度强于苯为配体形成的半三明治配合物, 取代苯形成的半三明治配合物之间进行比较, 随着F, Cl, Br取代的顺序逐渐增大, 与配体规律一致。
比较相同配体不同过渡金属离子形成的半三明治配合物的RM-环中心键长, 略有不同, Cr+, Mn2+形成的半三明治配合物的RM-环中心明显大于Ti2+, V2+形成的半三明治配合物, 约大0.3 Å , 考虑到配体相同, 影响其变化的主要因素应该是键级WBIC— M, Cr+, Mn2+形成的半三明治配合物的WBIC— M在0.03~0.1之间, 而Ti2+, V2+形成的半三明治配合物的WBIC— M在0.18~0.2之间, 明显增大了0.1左右, 键级大, 则键较强, 所以其键长较短。 不同配体相同过渡金属离子形成的半三明治配合物的RM-环中心键长之间相比较其大小顺序均符合: [(η 6-C6F6)M]n+> [(η 6-C6Cl6)M]n+> [(η 6-C6Br6)M]n+, 随着F, Cl, Br取代顺序, 金属离子到环中心的距离逐渐减小, 分析其变化原因, 金属离子与配体之间既存在静电引力, 又存在化学键即共价作用见表2, 随着F, Cl, Br取代的顺序, 除[(η 6-C6F6)V]2+, [(η 6-C6F6)Mn]2+特殊以外, 其余WBIC— M均逐渐增大, 增大幅度约0.01左右, 即RM-环中心随着键级的增大而减小, 分析其金属离子与碳环之间的静电引力发现, 氟苯为配体形成的半三明治配合物的VM-Ep碳环均为正值, 而其他配体形成的半三明治配合物的VM-Ep碳环均为负值, 即氟苯为配体形成的半三明治配合物中金属离子与碳环之间存在静电排斥力, 即具有强吸电子效应的F原子与C原子成键时, 诱导吸引了C上的电子, 导致C原子带正电荷, 以至于配体碳环整体带上正电荷, 与带正电荷的过渡金属离子相排斥, 导致以氟苯为配体的半三明治配合物中金属离子到环中心的距离最大; 而其他配体形成的半三明治配合物中金属离子和碳环之间存在静电引力, 因苯、 氯苯、 溴苯中H, Cl, Br原子与C原子成键时, C原子电负性强于前者, 自身吸引电子带上负电荷, 与带正电荷的过渡金属离子之间相互吸引, 随着F, Cl, Br取代的顺序其VM-Ep碳环由静电排斥力增大到静电引力, 导致金属离子到环中心的距离变短, 即RM-环中心随着静电引力的增大而减小。
为分析半三明治配合物的稳定性, 计算半三明治配合物的稳定化能[Δ E=E(半三明治配合物)-E(配体)-E(金属离子)], 发现所有半三明治配合物的稳定化能均小于零见表2, 其中相同配体不同金属离子构成的半三明治配合物的Δ E由小到大变化顺序为: [(η 6-C6X6)V]2+< [(η 6-C6X6)Mn]2+< [(η 6-C6X6)Ti]2+< [(η 6-C6X6)Cr]+, 稳定化能越小, 分子越稳定, 即Cr+形成的半三明治配合物最不稳定, 与其存在两个虚频, 不能稳定存在得到相同结论, 以取代苯为配体形成的半三明治配合物的Δ E的变化规律与[(η 6-C6H6)M]n+的Δ E的变化规律一致。 相同金属离子不同配体形成的半三明治配合物之间进行比较发现, 其Δ E由小到大变化顺序是: [(η 6-C6H6)M]n+< [(η 6-C6Br6)M]n+< [(η 6-C6Cl6)M]n+< [(η 6-C6F6)M]n+, 即氟苯为配体形成的半三明治配合物稳定性较差, 与其静电引力数据得到相同结论。
2.2.2 电子结构
初始设计、 计算了多种不同电子结构的半三明治配合物, 其中包括①金属原子(3d电子高自旋、 3d电子低自旋)、 ②金属离子即失去4s电子(3d电子高自旋、 3d电子低自旋)和③金属失去4s电子之后继续失去3d电子这样五种电子结构, 优化计算发现只有第②种情况中金属离子失去4s电子并且3d电子保持高自旋状态与苯、 取代苯形成的半三明治配合物能全部优化得到高对称性, 并且基本没有虚频, 与其他电子结构的半三明治配合物相比较能量较低(见表3), 说明该电子结构的半三明治配合物理论上可以存在, 而其他有些电子结构的半三明治配合物在理论上就是不能稳定存在的, 所以最终选择了金属原子失去4s电子且3d电子保持高自旋状态的金属离子与配体形成的半三明治配合物为研究对象。
| 表3 (η 6-C6X6)Mn2+的五种电子结构比较 Table 3 Compare five electronic structures for (η 6-C6X6)Mnn+ |
对第②种情况中金属失去4s电子并且3d电子保持高自旋状态与配体构成的半三明治配合物优化计算, 结果显示半三明治配合物确实是具有nA1(n=3, 4, 6)对称性的高自旋状态的分子(见表2)。
图3(a, b)是[(η 6-C6X6)Ti]2+(X=H, F)的部分特征分子轨道图, 其中[(η 6-C6H6)Ti]2+的HOMO-6, HOMO-11, [(η 6-C6F6)Ti]2+的HOMO-4和HOMO-18均是π 分子轨道诱导产生π 电子环流; 其中[(η 6-C6H6)Ti]2+的HOMO-16, [(η 6-C6F6)Ti]2+的HOMO-28是σ 分子轨道流诱导产生σ 电子环流, 均与配体相似既存在π 芳香性又存在σ 芳香性。 分析过渡金属离子与配体成键情况, 如[(η 6-C6H6)Ti]2+的HOMO-2是由Ti的3dxz轨道和C的2pz轨道相互作用形成的能量降低的δ 分子轨道, 其HOMO-3是由Ti的3dyz轨道和C的2pz轨道相互作用形成δ 分子轨道, 所以与配体分子不同之处在于半三明治配合物中存在三种类型的化学键即σ , π , δ 键, 其余半三明治配合物的成键情况与其类似, 不再一一例举。
 | 图3 [(η 6-C6H6)Ti]2+和[(η 6-C6X6)Ti]2半三明治配合物的特征分子轨道图 (a): [(η 6-C6H6)Ti]2+ ; (b): [(η 6-C6F6)Ti]2+Fig.3 Characteristic molecular orbitals for [(η 6-C6H6)Ti]2+and [(η 6-C6X6)Ti]2+ (a): [(η 6-C6H6)Ti]2+ ; (b): [(η 6-C6F6)Ti]2+ |
为了进一步探讨半三明治配合物芳香性, 分析其NBO电荷转移情况, 以下为[(η 6-C6X6)Ti]2+(X=H, F)中原子NBO电荷的自然排布:
[(η 6-C6H6)Ti]2+:
C [core]2s(0.99)2p(3.20[2
Ti [core]4s(0.06)3d(2.32[3
[(η 6-C6F6)Ti]2+
C [core]2s(0.85)2p(2.81[2
F [core]2s(1.82)2p(5.39[2
Ti [core]4s(0.05)3d(2.39[3
分析研究发现, 半三明治配合物的C, H, X的s轨道中电子数目均减小, 其2p轨道和金属离子的4p轨道、 3d轨道中电子数目均有所增加, [(η 6-C6H6)Ti]2+与苯相似, 存在σ 芳香性, 由C的2px, 2py参与形成, 如[(η 6-C6H6)Ti]2+的HOMO-6, 与配体相比较π 芳香性减小, σ 芳香性增大, Ti的4p轨道和3d轨道中电子分别增加了0.07和0.32e, 即[(η 6-C6H6)Ti]2+中C, H原子的s轨道中电子主要转移到了其C的2p轨道和Ti的3d轨道中; [(η 6-C6F6)Ti]2+与氟苯相似存在σ 芳香性, 由C的2px, 2py参与形成如[(η 6-C6F6)Ti]2+的HOMO-28, F的2pz增加0.88e主要是由其2px, 2py转移给2pz, F原子的2pz参与了大π 键的形成如[(η 6-C6F6)Ti]2+的HOMO-18, 与氟苯相比较π 芳香性减小, σ 芳香性增大, Ti的4p和3d轨道中电子分别增加了0.05和1.39e, 即[(η 6-C6F6)Ti]2+中C和F原子的2s轨道中电子主要转移到了其C的2p轨道和Ti的3d轨道中, 与[(η 6-C6H6)Ti]2+相比较π 芳香性增加, σ 芳香性减小。 氯苯与溴苯形成的半三明治配合物的π 芳香性与氟苯形成的半三明治配合物相似, σ 芳香性与苯形成的半三明治配合物相似。
总结发现相同金属离子不同配体形成的半三明治配合物中配体C, X的s电子转移至其2p轨道和金属3d轨道中电子数目由大到小的顺序是: [(η 6-C6F6)Ti]2+> [(η 6-C6Cl6)Ti]2+> [(η 6-C6Br6)Ti]2+, 而该顺序不仅与其配体电子转移顺序相一致, 与其NICS值描述的半三明治配合物的芳香性的变化顺序也相一致, 其他金属离子形成的半三明治与Ti2+有相似的规律不再一一举例。
2.3.1 配体芳香性
表1中NICS(0.0)为配体环中心处核独立化学位移值一般用来表征分子的σ 芳香性, NICS(1.0)为配体环中心上方1.0 Å 处核独立化学位移值一般用来表征分子的π 芳香性[7, 8, 10]。 所有配体的NICS值均为负值, 即配体分子均为芳香性分子。 其σ 芳香性由大到小的顺序是: (η 6-C6F6)> (η 6-C6Cl6)> (η 6-C6Br6)> (η 6-C6H6); 其π 芳香性由大到小的顺序是(η 6-C6F6)> (η 6-C6H6)> (η 6-C6Cl6)> (η 6-C6Br6), 该NICS(1.0)值的大小变化顺序与上述配体电子结构中p轨道中电子数目增加多少的顺序相一致, 充分证明了分子的π 芳香性主要由其p轨道贡献。
取代苯之间进行比较, 发现随着F, Cl和Br取代的顺序其NICS(0.0)值和NICS(1.0)值均增大, 芳香性逐渐减小, 即取代苯的芳香性顺序为(η 6-C6F6)> (η 6-C6Cl6)> (η 6-C6Br6)。
2.3.2 半三明治配合物芳香性分析
表2中所有半三明治配合物的NICS(0.0)值、 NICS(1.0)值(配体环中心正下方1.0 Å 处的核独立化学位移值)均为负值, 也是芳香性的物质。 与取代苯相比较, Ti2+和V2+形成的半三明治配合物的NICS值均小于配体的NICS值, 而Cr+和Mn2+形成的半三明治配合物的NICS值均大于配体的NICS值, 即相同取代苯为配体形成半三明治配合物的芳香性顺序: Ti2+> V2+> 配体> Cr+> Mn2+; 而苯及其形成的半三明治配合物的π 芳香性大小顺序与取代苯一致, 但是其σ 芳香性顺序中[(η 6-C6H6)V]2+> (η 6-C6H6)Ti]2+其余也与取代苯顺序一致。
相同金属离子与不同取代苯形成半三明治配合物的芳香性大小变化顺序与其配体变化顺序相一致[(η 6-C6F6)M]n+> [(η 6-C6Cl6)M]n+> [(η 6-C6Br6)M]n+, 即随着F, Cl和Br取代的顺序, 半三明治配合物的芳香性逐渐减小。
图1是配体(D6h)、 半三明治配合物(C6v)的A1g/A1对称类型的环伸缩振动模式, 计算配体(D6h)的全部对称操作, 求其可约表示Γ 总, 进行约化求出所含的不可约表示: Γ 振=A1g+B2g+2E2g+B1u+B2u+E1u+E2u。 根据光谱选律、 D6h点群的特征标表得出, x2+y2, z2都是不可约表示A1g的基, 所以A1g对称类型的正则振动是具有拉曼活性的, 即所有的配体分子均有拉曼活性。 同理分析半三明治配合物, 发现半三明治配合物(C6v)中均存在A1对称性的RSVRSF, 也具有拉曼活性, 与配体相似。
2.4.1 配体的环伸缩振动拉曼光谱频率
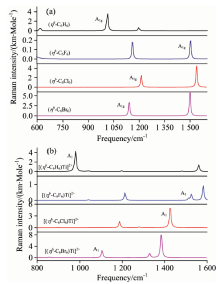
图4(a)是配体的RSVRSF理论计算值, 均存在明显的A1g对称类型的谱峰, 且强度很高, 说明配体分子的A1g对称类型的RSVRSF在实验上是能够测量的, 而且本工作前期对苯的RSVRSF的实验值进行了测量, 实验值与理论计算值基本一致[12]。 其频率数值由大到小的变化顺序是(η 6-C6F6)> (η 6-C6Cl6)> (η 6-C6Br6)> (η 6-C6H6), 即取代苯的RSVRSF均大于苯, 并且随着F, Cl, Br取代的顺序逐渐减小。 分析其频率变化原因与力常数相关, 即力常数越大其振动频率就越大, 其中氟苯、 氯苯、 溴苯和苯的RSVRSF对应的力常数分别是17.044, 10.668, 9.923, 3.643见表1, 逐渐减小。
 | 图4 (η 6-C6X6)和[(η 6-C6X6)Ti]2+(X=H, F~Br)的理论计算拉曼光谱图 (a): (η 6-C6X6)(X=H, F~Br); (b): [(η 6-C6X6)Ti]2+(X=H, F~Br)Fig.4 Theoretical calculation Raman spectrum for (η 6-C6X6) and [(η 6-C6X6)Ti]2+(X=H, F~Br) (a): (η 6-C6X6)(X=H, F~Br); (b): [(η 6-C6X6)Ti]2+(X=H, F~Br) |
2.4.2 半三明治配合物的环伸缩振动拉曼光谱频率
图4(b)是[(η 6-C6X6)Ti]2+(X=H, F~Br)环伸缩振动拉曼光谱频率(RSVRSF)的理论计算值, 均存在明显的A1对称类型的谱峰, 并且强度很高, 说明半三明治配合物的A1对称类型RSVRSF在实验上也是能够测量的。 其他半三明治配合物的理论计算拉曼光谱图与之相似, 不再一一列举。
与配体比较, 形成半三明治配合物后, 其RSVRSF值变化较小见表1。 但相同配体和不同金属离子形成的半三明治配合物之间比较, [(η 6-C6H6)M]n+(M=Ti~Mn, n=1, 2)的四个半三明治配合物的A1对称类型的RSVRSF值分别为978.67, 981.66, 988.04和975.70 cm-1, 均小于配体(η 6-C6H6)的RSVRSF值1 012.83 cm-1, 即在(η 6-C6H6)上方加入过渡金属离子形成半三明治配合物后, 其RSVRSF值相比较于(η 6-C6H6)有所减小, 分析原因发现金属离子与配体C原子之间不仅存在共价作用力, 还存在一定的静电引力(见表2), 导致RSVRSF值有所减小, 但是减小的幅度不大, 约30 cm-1; [(η 6-C6F6)M]n+(M=Ti~Mn, n=1, 2)中四个半三明治配合物的A1对称类型的RSVRSF值分别为1 527.23, 1 533.52, 1 503.15和1 521.54 cm-1, 均大于(η 6-C6F6)的RSVRSF值1 503.15 cm-1, 即在配体(η 6-C6F6)上方加入过渡金属离子形成半三明治配合物后, 其RSVRSF值相比较于(η 6-C6F6)有所增大, 分析原因发现金属离子与配体C原子之间存在共价作用力, 还存在一定的静电排斥力(见表2), 导致RSVRSF值有所增大, 增大约20.0 cm-1; 以氯苯、 溴苯为配体形成的半三明治配合物的变化规律与[(η 6-C6H6)M]n+相似, 是其力常数、 键级、 静电引力三者综合作用的结果。
相同金属离子不同配体形成半三明治配合物的RSVRSF与其配体的RSVRSF变化规律一致, 即其RSVRSF由大到小的顺序是: [(η 6-C6F6)Ti]2+> [(η 6-C6Cl6)Ti]2+> [(η 6-C6Br6)Ti]2+> [(η 6-C6H6)Ti]2+。 取代苯形成的半三明治配合物的RSVRSF大于苯形成的半三明治配合物, 而且随着F, Cl和Br取代顺序逐渐减小, 同配体变化规律相同。 同理分析其原因, 与力常数有关, 即力常数越大其RSVRSF越大, 还应该与C— C键级、 C— M键级有关, 考虑取代原子的变化发现, 电负性大的原子诱导吸引了环上C原子的电子, C原子之间成键变强, 所以C原子之间伸缩振动的频率变大, 即[(η 6-C6F6)Ti]2+的RSVRSF最大, 而[(η 6-C6H6)Ti]2+的最小。
物质之所以表现出芳香性(π 芳香性、 σ 芳香性)是因分子内部存在π 和σ 电子环流, 如配体的特征分子轨道图(η 6-C6H6)的HOMO-4和HOMO-14, (η 6-C6F6)的HOMO-2和HOMO-14(见图2), 半三明治配合物的特征分子轨道图[(η 6-C6H6)Ti]2+的HOMO-6和HOMO-16, [(η 6-C6F6)Ti]2+的HOMO-4, HOMO-18和HOMO-28(见图3), 电子环流的形成是因为分子内部存在共轭键, 这种共轭键是由分子内原子间大小相等的共轭作用力所决定, 这种共轭作用力表现出的振动方式即为环伸缩振动, 所以理论上来讲, 物质的芳香性与分子的环伸缩振动存在一定的相关性; 然而分子的环伸缩振动有很多种类型, 通过群论分析发现分子A1g/A1对称类型的环伸缩振动具有很强的拉曼光谱活性, 因此理论上分子的芳香性与其A1g/A1对称类型的环伸缩振动拉曼光谱频率存在一定的相关性。 下面具体讨论配体、 半三明治配合物的RSVRSF与其芳香性(NICS)的相关性, 见图5和图6。
2.5.1 配体环伸缩拉曼光谱大小与其芳香性(NICS)大小相关性
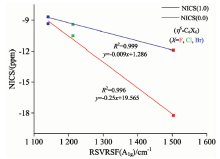
图5为配体A1g对称类型的RSVRSF值与NICS值的相关性曲线, 通过以上对配体芳香性、 拉曼光谱频率分析可知, 分子的RSVRSF越大其芳香性也越大。 其中配体的RSVRSF与其NICS(0.0)的相关系数达到0.999, 单侧检验的p值为0.020、 双侧检验的p值为0.041两者均小于0.05, 说明配体RSVRSF与其NICS(0.0)之间的相关显著性; 同理配体的RSVRSF与其NICS(1.0)的相关性达到0.996, 单侧检验的p值为0.008、 双侧检验的p值为0.017两者也均小于0.05, 说明配体分子的RSVRSF与其NICS(1.0)之间的相关同样显著。 说明取代苯的A1g对称类型的RSVRSF与其NICS(0.0)和NICS(1.0)均存在高度的相关性, 认为理论上可以通过测量取代苯的A1g对称类型的RSVRSF大小来测量其芳香性大小, 为实验测量取代苯芳香性大小提供新的实验方法和理论依据。
 | 图5 (η 6-C6X6)(X=F~Br)RSVRSF(A1g)(cm-1)与NICS(ppm)的相关性曲线Fig.5 The correlation curve between the RSVRSF (A1g) (cm-1) values and NICS(ppm) for (η 6-C6X6)(X=F~Br) |
2.5.2 半三明治配合物环伸缩拉曼光谱强弱与其芳香性(NICS)的相关性
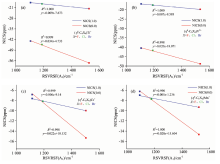
图6是取代苯为配体形成的半三明治配合物的A1对称类型的RSVRSF值与其NICS值的相关性曲线, 与配体规律相似。 半三明治配合物的芳香性NICS值与其RSVRSF值也呈一定的正相关。 如[(η 6-C6X6)Ti]2+(X=F~Br)系列RSVRSF与其NICS(0.0)的相关性达到0.999, 单侧检验的p值为0.011、 双侧检验的p值为0.022两者均小于0.05, 说明[(η 6-C6X6)Ti]2+的RSVRSF与其NICS(0.0)之间显著相关, 其RSVRSF与NICS(1.0)的相关性达到1.000, 单侧检验的p值为0.002、 双侧检验的p值为0.004两者也均小于0.05, 说明[(η 6-C6X6)Ti]2+的RSVRSF与其NICS(1.0)之间也是显著相关的; 其他系列半三明治配合物规律与[(η 6-C6X6)Ti]2+(X=F~Br)相似。 说明半三明治配合物的RSVRSF与其NICS的相关性均是显著的, 即半三明治配合物的RSVRSF越大其芳香性越强。 认为理论上可以通过测量取代苯形成的半三明治配合物的A1对称类型的RSVRSF大小来测量其芳香性强弱, 为实验测量以取代苯为配体形成的半三明治配合物的芳香性大小提供新的实验方法和理论依据。
 | 图6 [(η 6-C6X6)M]n+(X=F~Br, M=Ti~Mn, n=1, 2)的RSVRSF(A1g)(cm-1)与芳香性(NICS)(ppm)相关性曲线 (a): [(η 6-C6X6)Ti]2+; (b): [(η 6-C6X6)V]2+; (c): [(η 6-C6X6)Cr]+; (d): [(η 6-C6X6)Mn]2+Fig.6 The correlation curve between the RSVRSF (A1g) (cm-1) values and NICS(ppm) for [(η 6-C6X6)M]n+(X=F~Br, M=Ti~Mn, n=1, 2) (a): [(η 6-C6X6)Ti]2+; (b): [(η 6-C6X6)V]2+; (c): [(η 6-C6X6)Cr]+; (d): [(η 6-C6X6)Mn]2+ |
(η 6-C6X6)(X=F~Br)均属于D6h点群, 且均无虚频为稳定结构, 半三明治配合物均属于C6v点群, 其中[(η 6-C6X6)Cr]+系列均有两个虚频为不稳定结构, [(η 6-C6Br6)Ti]2+有一个虚频为过渡态, 其余分子均没有虚频为稳定结构。 所有目标分子的NICS值均为负值, 均为芳香性分子。 配体中均存在A1g对称性的RSVRSF, 半三明治配合物中则均存在A1对称性的RSVRSF, 拉曼光谱强度都很大, 取代苯及其形成的半三明治配合物的NICS的绝对值、 RSVRSF值均随着F, Cl和Br取代的顺序逐渐减小, 呈高度正相关, 且相关性系数均达到0.99以上。 理论预测可通过在实验上定量测定目标分子的A1g/A1对称性的RSVRSF大小实现对其芳香性大小的定量测定, 分子的RSVRSF越大, 其芳香性越强。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|