
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TLC-SERS联用快速同时检测食品中非法添加的碱性橙Ⅱ和酸性橙Ⅱ的研究
[符云鹏 , 齐颖, 扈晓鹏, 佟蕊, 方国臻
, 齐颖, 扈晓鹏, 佟蕊, 方国臻* , 王硕]
, 齐颖, 扈晓鹏, 佟蕊, 方国臻, 王硕]
|
|


作者简介: 符云鹏, 1989年生, 天津科技大学食品工程与生物技术学院硕士研究生 e-mail: yunpeng_ling@126.com
建立了一种薄层色谱(TLC)与表面增强拉曼光谱(SERS)联用快速检测食品中非法添加的碱性橙Ⅱ和酸性橙Ⅱ的方法。 采用薄层色谱法对样品提取液进行简单分离, 并优化了薄层色谱分离条件; 合成并优选出水相和有机相两类银溶胶, 分别用作碱性橙Ⅱ和酸性橙Ⅱ拉曼信号增强基底, 继而利用优选的银溶胶为增强基底对分离出的微量物质进行表面增强拉曼光谱检测, 考察了检测时间, 并确立了碱性橙Ⅱ和酸性橙Ⅱ检出限分别为1和2.5 mg·L-1。 将该方法用于实际样品检测, 成功实现了复杂食品基质中碱性橙Ⅱ和酸性橙Ⅱ的同时快速检测。 该方法具有简便、 快速、 经济、 专属性好等优势, 为复杂食品基质中碱性橙Ⅱ和酸性橙Ⅱ的同时快速检测提供了新方案。
A method for the rapid detection of illegally added basic orange Ⅱ and acid orange Ⅱ in food was established by the combination of thin layer chromatography (TLC) and surface enhanced Raman spectroscopy (SERS). TLC was used for the separation of sample extracts tentatively and the conditions were optimized. Two kinds of silver sols, aqueous phase and organic phase, were synthesized and used as the substrate to enhance Raman signal for basic orange Ⅱ and acid orange Ⅱ. SERS was used to detect isolated micro-substances and the time was investigated. The detection limits of basic orange Ⅱ and acid orange Ⅱ were 1 and 2.5 mg·L-1. Then, the method has been successfully used for rapid detection of basic orange and orange Ⅱ in the complex food matrix. The method has the advantages of simplicity, rapidity, economic and specificity, and will provide a new scheme for the simultaneous detection of basic orange Ⅱ and acid orange Ⅱ in food.
碱性橙Ⅱ 和酸性橙Ⅱ 是一类偶氮类工业染料, 已证实人体直接接触有一定致病性, 中等毒性和致癌性[1]。 但由于其具有易上色, 成本低等特点, 被一些不法商贩用于豆制品等食品的染色, 危害了消费者的身体健康。 目前报道碱性橙Ⅱ 和酸性橙Ⅱ 同时检测采用的是液相色谱-串联质谱法[2]。 但样品前处理过程繁杂耗时, 仪器昂贵, 不能做到现场快速检测。 将薄层色谱(thin layer chromatography, TLC)与表面增强拉曼光谱(surface-enhanced Raman spectroscopy, SERS)联用, 利用TLC简单分离复杂体系后, 再利用SERS获得样品体系中微量待检成分的拉曼光谱信息进行分析, 是一种很好的检测鉴别方法。 目前这种技术已被用于多种分析物的分析和检测, 如用于活体生物样品中可替宁、 吗啡的检测[3, 4]、 环境中芳香族污染物的检测[5]、 药物中有效成分和非法添加物的分析鉴别[6]。 但作为一种食品安全检测方法并未见广泛研究与报道。
本文建立了薄层色谱与表面增强拉曼光谱联用快速同时检测食品中非法添加染料碱性橙Ⅱ 和酸性橙Ⅱ 的方法, 对薄层色谱分离条件、 表面增强拉曼光谱检测条件进行了优化, 并用于实际样品检测, 为复杂食品基质中碱性橙Ⅱ 和酸性橙Ⅱ 的同时快速检测提供了新方案。
显微共聚焦拉曼光谱仪(InVia, 英国雷尼绍Renishaw公司); ZF-7型暗箱三用紫外分析仪; KH-300DB型超声波清洗器。
碱性橙Ⅱ 标准品(99%); 酸性橙Ⅱ 标准品(99%); 硝酸银(AR); 柠檬酸钠(AR); 优级纯N, N-二甲基甲酰胺 (DMF); 聚乙烯比咯烷酮 (PVPK40); 其他试剂均为分析纯; HSGF254薄层板(25 mm× 75 mm)。
1.2.1 水相银溶胶
将浓度为0.02% AgNO3溶液150 mL放入微波炉中, 微波火力开置最大, 加热5 min, 溶液出现沸腾后, 从微波炉中取出, 迅速加入4 mL浓度为1%柠檬酸钠溶液, 放入微波炉中再加热2 min, 溶液沸腾, 取出冷却备用[7]。
1.2.2 有机相银溶胶
将10 mL AgNO3与PVP K40(摩尔比10:1)的混合溶液加入到100 mL沸腾的DMF中, 其中AgNO3浓度为 1× 10-3 mol· L-1, 继续加热1 min, 取出冷却备用[8]。
1.3.1 标准品溶液配制
准确称取碱性橙Ⅱ 和酸性橙Ⅱ 标准品各10.0 mg于100 mL容量瓶中, 甲醇溶解并定容, 混匀, 作为标准品溶液备用。
1.3.2 模拟阳性样品制备及提取方法
模拟阳性样品制备: 称取1.00 g粉碎后样品多份置于离心管中, 加入碱性橙Ⅱ 和酸性橙Ⅱ 混合标准溶液, 使碱性橙Ⅱ 、 酸性橙Ⅱ 添加量分别相当于10, 6和3 mg· kg-1, 振荡15 min后混匀, 放置过夜。
提取方法: 加入5 mL 8%氨水乙腈溶液涡旋后超声提取25 min, 加入5 mL正己烷, 涡旋, 静置弃去上层正己烷, 4 000 r· min-1离心15 min, 转移上清液至另一只离心管中, 重复提取一次, 合并提取液, 移至试管内35 ℃下氮吹至近干, 甲醇复溶至1 mL。
将薄层板置于光谱仪显微镜载物台上, 激光光斑聚焦到样品上, 对样品斑点进行光谱采集。 采用激光光源(λ =785 nm), 碱性橙Ⅱ 斑点光谱仪检测激光功率2.5 mW, 酸性橙Ⅱ 斑点光谱仪检测激光功率为500 mW, 都选用50倍物镜, 单次曝光时间20 s, 信号累计2次, 采集范围400~2 000 cm-1, 重复多次, 选用信号噪声比(signal-to-noise ratio, SNR)高光谱求平均。
吸取标准品和模拟阳性样品溶液各6 μ L, 点样于同一薄层板上不同点位, 展开取出, 晾干后, 标准品点位直接检视定位, 定位阳性样品色谱条带中与标准品Rf值相同的位置, 滴加聚集剂和银溶胶, 用光谱仪对锁定点位进行光谱采集, 将所得光谱与标准品光谱对比分析。
用拉曼光谱仪自带的Manual WiRE3.4软件采集光谱及预处理, 采用Origin 8.5软件处理数据和作图。
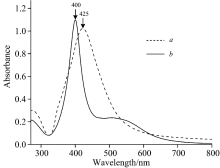
对两类银溶胶进行UV-Vis光谱扫描和透射电镜表征, 光谱扫描范围: 250~800 nm。 从图1(a)可以看出, 水相银溶胶只有425 nm一个明显的吸收峰, 结合图2(a), 可知银纳米粒子多数为类球型结构, 其中有少数为棒状粒子, 粒径主要分布在35~55 nm之间; 从图1(b)可以看出, 有机相银溶胶也只有400 nm一个最大吸收峰, 而且半峰宽较窄, 说明纳米颗粒粒径均匀, 结合图2(b), 可知银溶胶粒子大部分为类球型, 其粒径主要分布在40 nm左右。
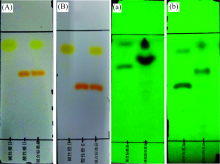
选用正丁醇-乙醇-1%氨水(6:2:2)作为展开剂[9], 效果如图3(A)所示, 展开后碱性橙Ⅱ 和酸性橙Ⅱ 能够很好的分离, 但两者的Rf值都偏大。 在后续食品样品实验中, 调整正丁醇和乙醇比例, 使之比例变为(10:1:2), 展开后不仅两种待测物能够分开, 且Rf值有所降低, 如图3(B)所示, 而且从两种展开剂的腐竹、 油豆皮及卤蛋混合提取液展开图[图3(A)和(B)]可以看出, 后者两种待检目标物与样品混合提取液残余基质具有更好的分离效果, 起到净化作用。
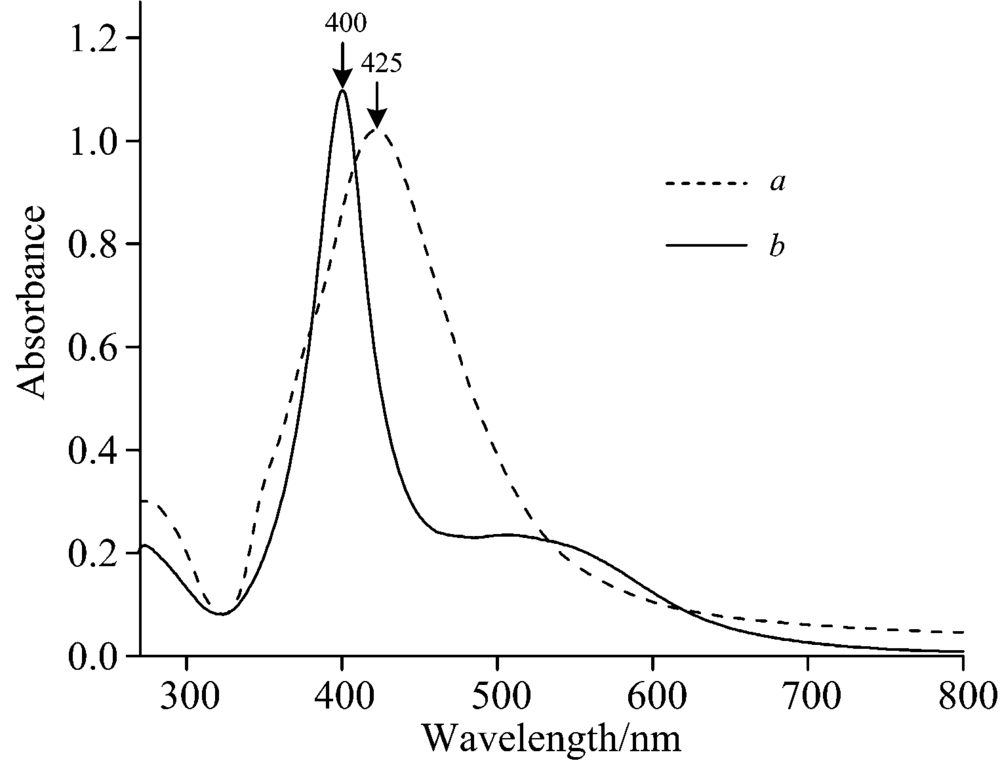 | 图1 水相和有机相银溶胶UV-Vis吸收图(a): 水相; (b): 有机相Fig.1 UV-Vis absorption spectra of aqueous and organic silver sols(a): Water; (b): DMF |
 | 图2 水相和有机相银溶胶TEM图(a): 水相; (b): 有机相Fig.2 TEM of aqueous and organic silver sol(a): Water; (b): DMF |
 | 图3 标准品和食品样品的薄层色谱展开图Fig.3 TLC expanded view of standard and food samples |
尝试柠檬酸钠还原制备的水相银溶胶做薄层板表面碱性橙Ⅱ 拉曼信号增强基底, 点胶量为2.5 μ L, 不加聚集剂之前, 拉曼信号增强效果很微弱。 但在滴加银溶胶之前, 先滴加2.5 μ L, 0.01%NaCl溶液做聚集剂之后再次检测, 获得了优质的碱性橙Ⅱ 表面增强拉曼光谱。
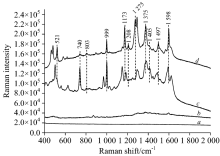
如图4a所示, 薄层板水相银溶胶背景拉曼光谱没有很强信号峰。 从图4b和c比较中可以看出, 与同浓度的常规拉曼光谱相比, 表面增强拉曼光谱中碱性橙Ⅱ 大部分拉曼特征峰都能够在混合光谱中显现, 且信号强度有显著增强。 但并不是全部均匀的增强, 部分拉曼峰增强效果明显, 例如704, 999, 1 173, 1 208, 1 275, 1 375, 1 405, 1 497和1 598 cm-1, 有的则相对较微弱521和803 cm-1, 另外704 cm-1处峰强则处于不稳定状态, 这可与跟薄层板中某些成分有关, 另外还有一些峰发生了波数偏移, 例如1 173和1 208 cm-1发生了蓝移, 1 275 cm-1发生了红移, 推测可能是碱性橙Ⅱ 结构有所变化, 或者增强时改变了其峰的原本的振动规律。 此外999, 1 173, 1 375和1 598 cm-1这四个分散特征峰在光谱中能够清晰辨别出来, 信号强度较高, 可以作为碱性橙Ⅱ 表面增强拉曼检测中的定性特征峰。
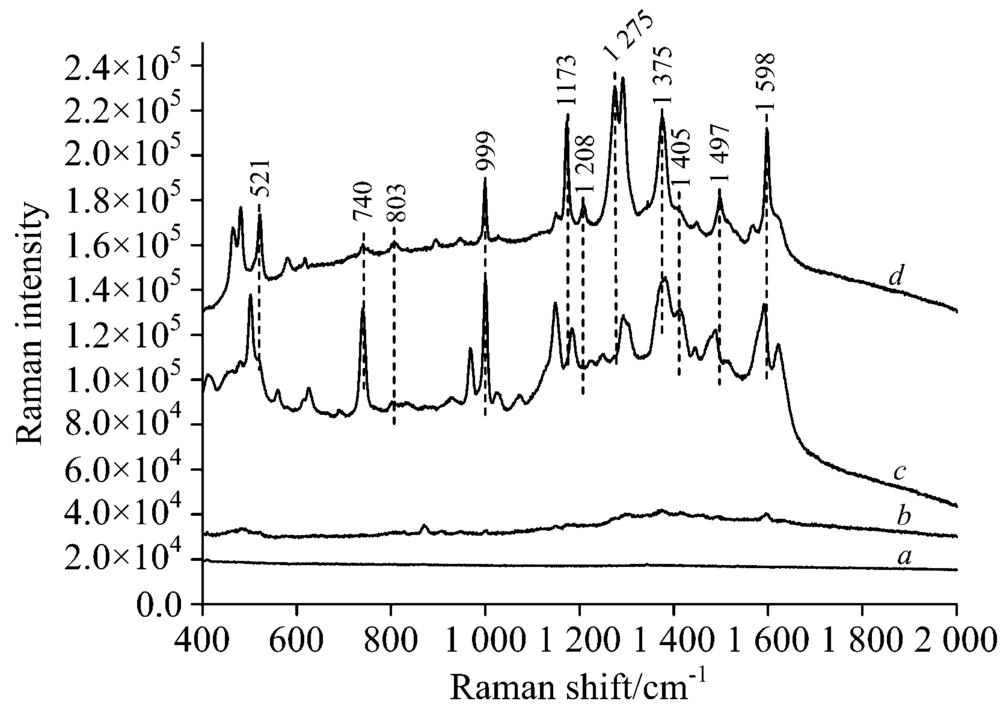 | 图4 a薄层板银溶胶背景拉曼光谱; b 25 mg· L-1碱性橙Ⅱ TLC常规拉曼光谱; c 25 mg· L-1碱性橙Ⅱ TLC-SERS光谱; d 载玻片表面100 mg· L-1碱性橙Ⅱ 常规拉曼光谱Fig.4 a Raman Spectra of silver sol on the TLC; b Raman spectrum of the 25 mg· L-1 basic orange Ⅱ separated by TLC; c SERS of the 25 mg· L-1 basic orange Ⅱ separated by TLC; d Raman spectra of 100 mg· L-1 basic orange Ⅱ on the glass plate |
尝试用柠檬酸钠还原制备不同粒径的水相银溶胶去做薄层色谱板表面酸性橙Ⅱ 拉曼信号增强基底, 都没有发现明显的增强效果, 尝试滴加常用聚集剂, 效果仍不理想。 考虑到酸性橙Ⅱ 负电性可能弱于柠檬酸根离子, 采用麦芽糖还原制备水相银溶胶去做增强, 效果也较差。 猜测可能由于酸性橙Ⅱ 吸附在薄层板内层, 难以与水溶性的银粒子结合形成SERS“ 热点” , 故无法产生较好的增强效果。 因此尝试采用DMF做溶剂制备有机相银溶胶去做增强基底, 点胶量为2.5 μ L, 获得了优质的酸性橙Ⅱ 表面增强拉曼光谱。
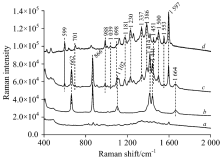
从图5b可以看出薄层板银溶胶的背景光谱有6个拉曼峰分别是662, 866, 1 102, 1 411, 1 441和1 664 cm-1, 这些拉曼峰来自银溶胶中溶剂DMF。 实际检测时, 待测物的拉曼光谱谱中仍存在这些背景峰, 但这并未影响待测物本身的拉曼信号。 从图5c和d可以看出SERS光谱中拉曼信号峰基本上是背景拉曼峰和酸性橙Ⅱ 拉曼峰的叠加, 再将其与图5a对比可以看出, 与同浓度的常规拉曼光谱相比, 表面增强拉慢光谱中酸性橙Ⅱ 大部分拉曼特征峰都能够在混合光谱中显现, 且信号强度有显著增强, 增强后的酸性橙Ⅱ 特征峰没有像碱性橙Ⅱ 一样发生大幅波数偏移, 各个拉曼峰增强效果也比较均匀。 从中可以清晰看到酸性橙Ⅱ 特征峰: 599, 701, 988, 1 181, 1 230, 1 337, 1 386, 1 500, 1 553和1 597 cm-1, 其中1 597 cm-1特征是所有特征峰中相对强度最强的。 此外988, 1 230, 1 337和1 597 cm-1这四个分散特征峰在光谱中能够清晰辨别出来, 信号强度较高, 也无背景信号干扰, 可以作为酸性橙Ⅱ 表面增强拉曼检测特征峰。
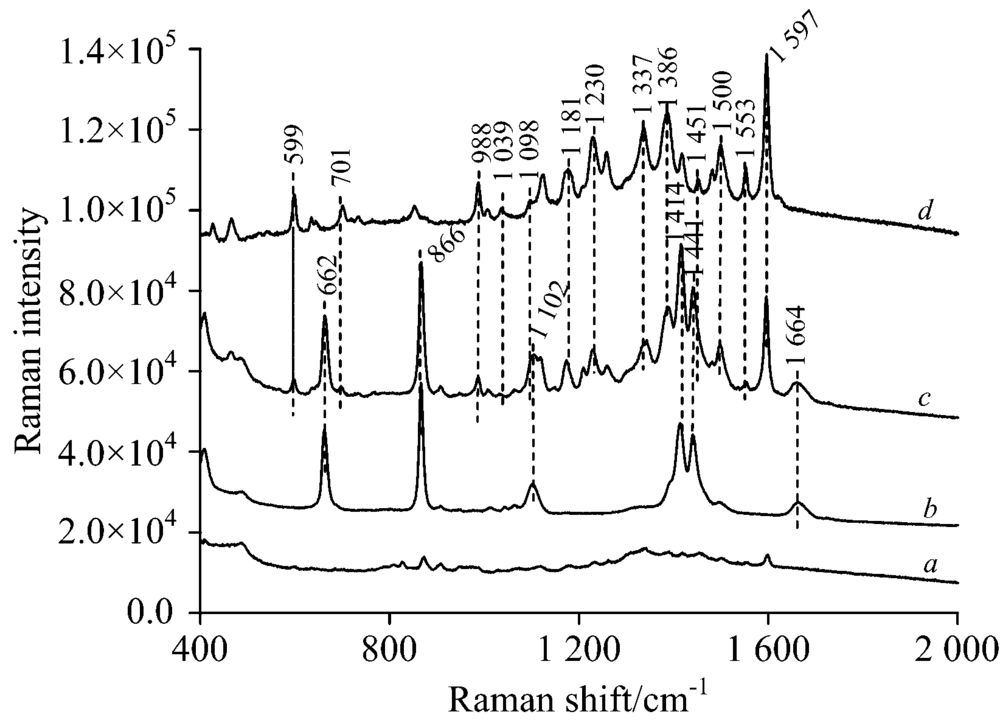 | 图5 a 25 mg· L-1酸性橙Ⅱ TLC常规拉曼光谱; b薄层板银溶胶背景拉曼光谱; c 25 mg· L-1酸性橙Ⅱ TLC-SERS光谱; d载玻片表面100 mg· L-1酸性橙Ⅱ 常规拉曼光谱Fig.5 a Raman spectrum of the 25 mg· L-1 acid orange Ⅱ separated by TLC; b Raman Spectra of silver sol on the TLC; c SERS of the 25 mg· L-1 acid orange Ⅱ separated by TLC; d Raman spectra of 100 mg· L-1 acid orange Ⅱ on the glass plate |
表面增强拉曼光谱的质量与采集时间有一定相关性, 以25 mg· L-1碱性橙Ⅱ 和酸性橙Ⅱ 标准溶液作为点样液, 按照1.5节对SERS光谱质量影响因素检测时间进行考察。
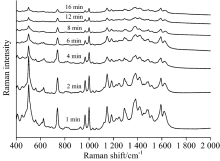
从图6可以看出, 在聚集剂和未浓缩水相银溶胶滴加后, 碱性橙Ⅱ 拉曼信号强度是一个逐渐减弱的过程。 在滴加1 min后, 拉曼信号增强效果最好。 随着时间的延长, 拉曼信号强度逐渐减弱, 8 min后拉曼信号强度也趋于不变。 银溶胶滴加后, 大量的水在薄层色谱板表面形成一个薄的水膜, 沉积在薄层板内的碱性橙Ⅱ 分子重新溶解并吸附在银粒子表面, 和银纳米粒子一起悬浮于水膜里, 此时检测能获得高信号强度拉曼光谱。 随着水分的挥发, 水膜逐渐变薄消失, 水中基底与碱性橙Ⅱ 分子接触机会减小, 单位基底吸附的碱性橙Ⅱ 分子减少, 拉曼信号增强效果逐渐减弱, 待水分挥发完全, 检测体系变为干斑, 拉曼信号则趋于稳定。
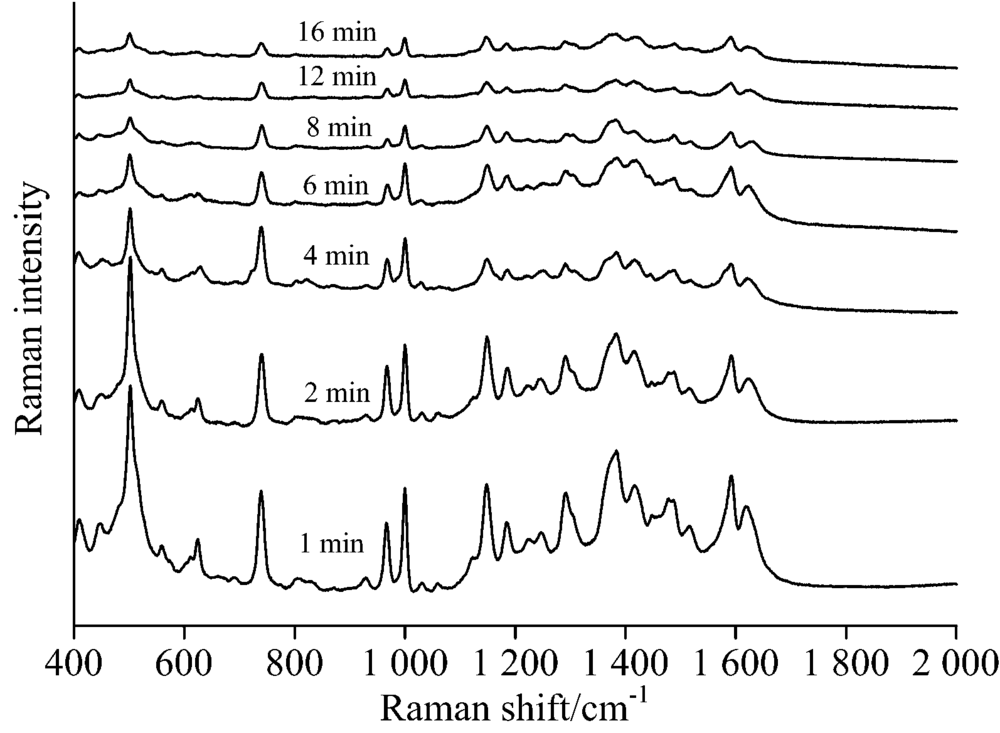 | 图6 不同时间碱性橙Ⅱ TLC-SERS光谱Fig.6 SERS of basic orange Ⅱ at different time separated by TLC |
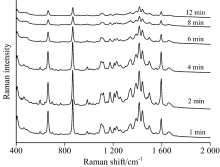
采集滴加未浓缩有机相银胶后不同时间酸性橙Ⅱ 表面增强拉曼光谱, 如图7所示。 在滴加银溶胶后立即进行检测, 所得的拉曼光谱含有很强的背景信号, 以背景峰866 cm-1强度可以看出滴加银胶后背景信号最强, 之后随着时间推移开始逐渐减弱; 而待测物信号强度却与之不同, 从1 597 cm-1可以看出酸性橙Ⅱ 峰强是一个先增大后减弱的趋势, 在滴加银溶胶1 min后就可以观察到很明显的酸性橙Ⅱ 表面增强拉曼信号, 当时间推移到2 min后其拉曼信号强度进一步增大, 之后开始减弱。 随着时间继续向后推移, 两者信号都开始减弱, 8 min后达到相对稳定的状态。 由图可知滴加银胶2 min后采集拉曼光谱较合适, 此时待测物拉曼信号强度与背景峰强度相对比值最大, 且待测物拉曼信号较丰富且峰较强。
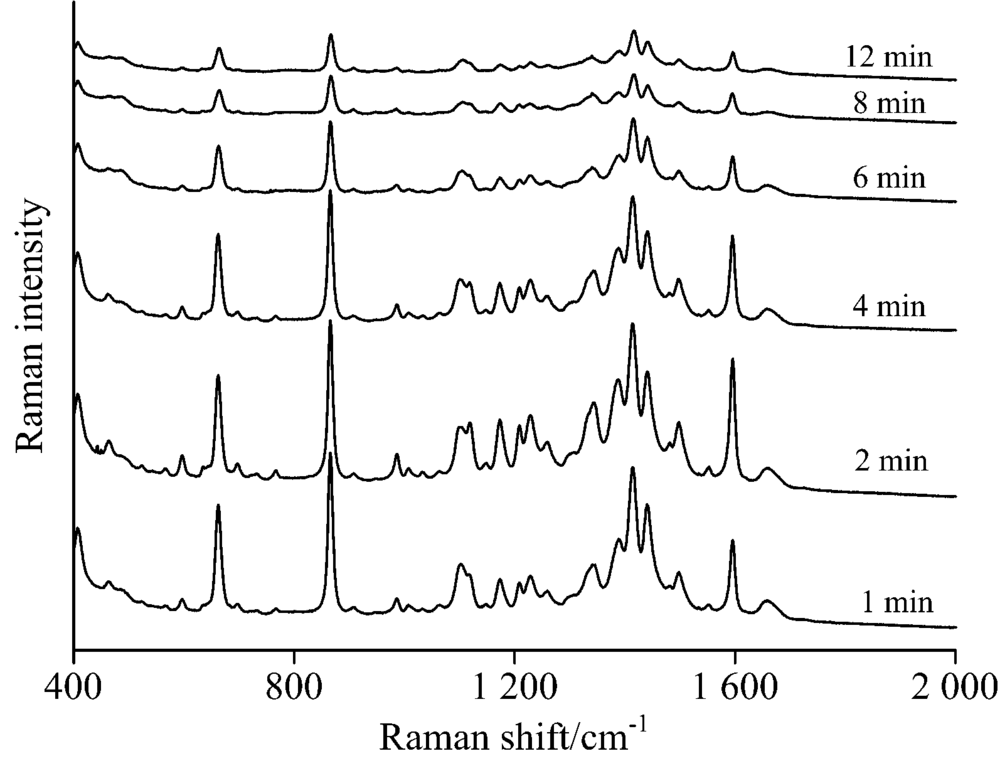 | 图7 不同时刻酸性橙Ⅱ TLC-SERS光谱Fig.7 SERS of acid orange Ⅱ at different time separated by TLC |
综上所述, 碱性橙Ⅱ 在分别滴加2.5 μ L, 0.1% NaCl溶液和水相银溶胶1 min后进行SERS检测, 酸性橙Ⅱ 滴加2.5 μ L有机相银溶胶2 min后进行SERS检测可获得较理想的效果。
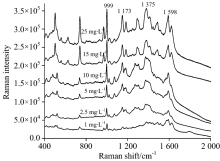
将碱性橙Ⅱ 和酸性橙Ⅱ 标准溶液稀释为25, 15, 10, 2.5和1 mg· L-1的混合溶液, 将上述混合溶液与25 mg· L-1混合溶液一起按照1.5节提供检测方法点样于同一张薄层板的不同点位, 在上述实验中获得的碱性橙Ⅱ 和酸性橙Ⅱ SERS最佳检测条件下, 在设定好的检测仪器参数下, 浓度从高到低, 依次采集薄层板表面锁定待测物点位的SERS光谱, 直至采集到的光谱中待测物特征峰无法辨认为止。 采集到的碱性橙Ⅱ 的点位的表面增强拉曼光谱如图8所示, 当标准品溶液浓度低至1 mg· L-1, 依然可以清晰看到999, 1 173, 1 375和1 598 cm-1这四个碱性橙Ⅱ 的特征峰。 当标准品溶液浓度低于1 mg· L-1时, 待测物特征峰不清晰, 确认其检出限为1 mg· L-1, 其对应的点样沉积量仅为0.006 μ g, 明显低于其在紫外-可见灯下的检出限, 与常规拉曼光谱相比光谱信噪比高, 检出限低。
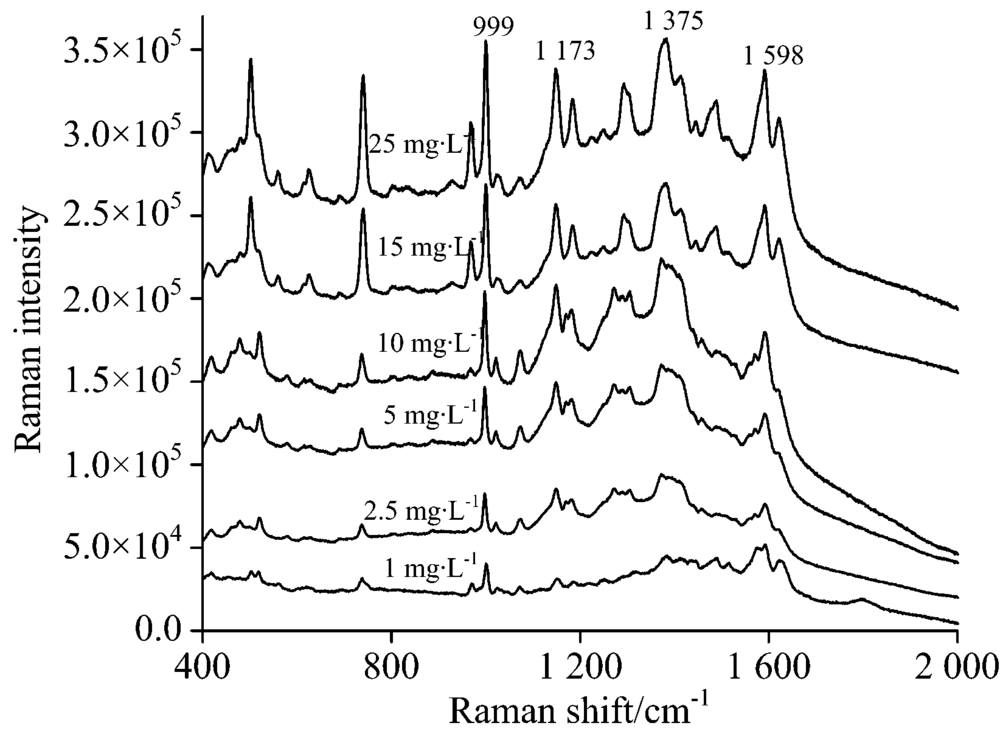 | 图8 不同浓度碱性橙Ⅱ TLC-SERS光谱Fig.8 SERS of basic orange Ⅱ at different concentrations separated by TLC |
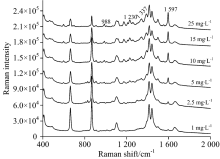
采集到的酸性橙Ⅱ 的点位的表面增强拉曼光谱如图9, 当标准品浓度低至2.5 mg· L-1, 酸性橙Ⅱ 的薄层板表面增强拉曼光谱中定性检测的四个特征峰988, 1 230, 1 337和1 597 cm-1, 除1 597 cm-1依然可以清晰看见外, 其他特征峰由于背景信号干扰已经无法辨别出。 所以确认其检出限为2.5 mg· L-1, 其对应的点样沉积量仅为0.015 μ g, 显著低于其在紫外-可见灯下的检出限, 且与常规拉曼相比光谱信噪比高, 检出限低。
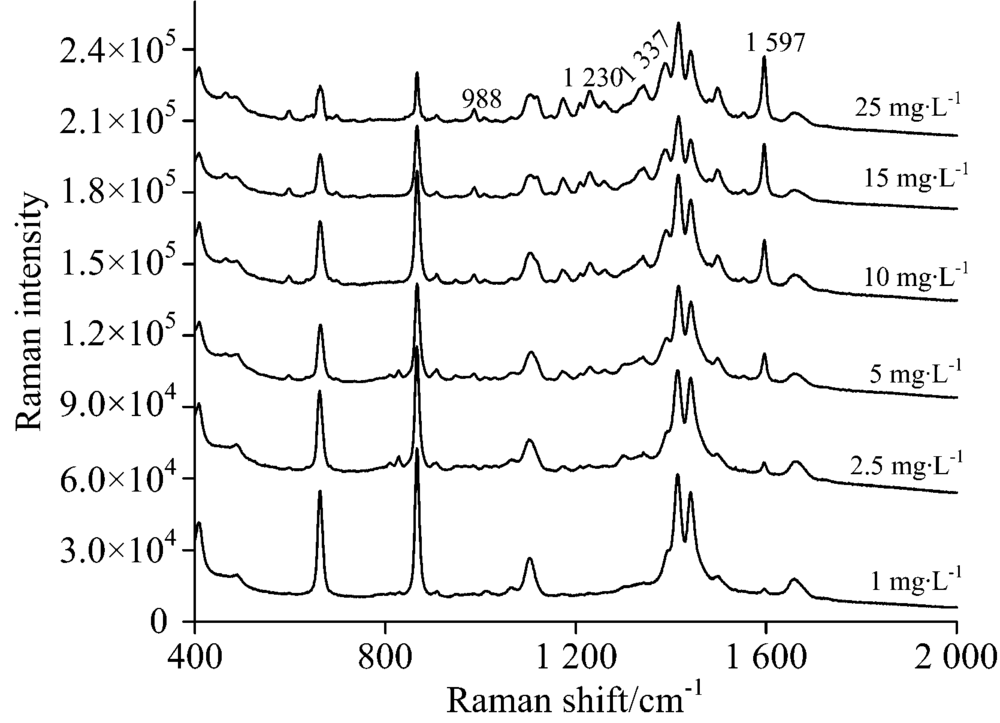 | 图9 不同浓度酸性橙Ⅱ TLC-SERS光谱Fig.9 SERS of acid orange Ⅱ at different concentrations separated by TLC |
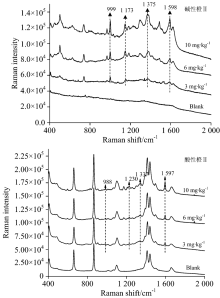
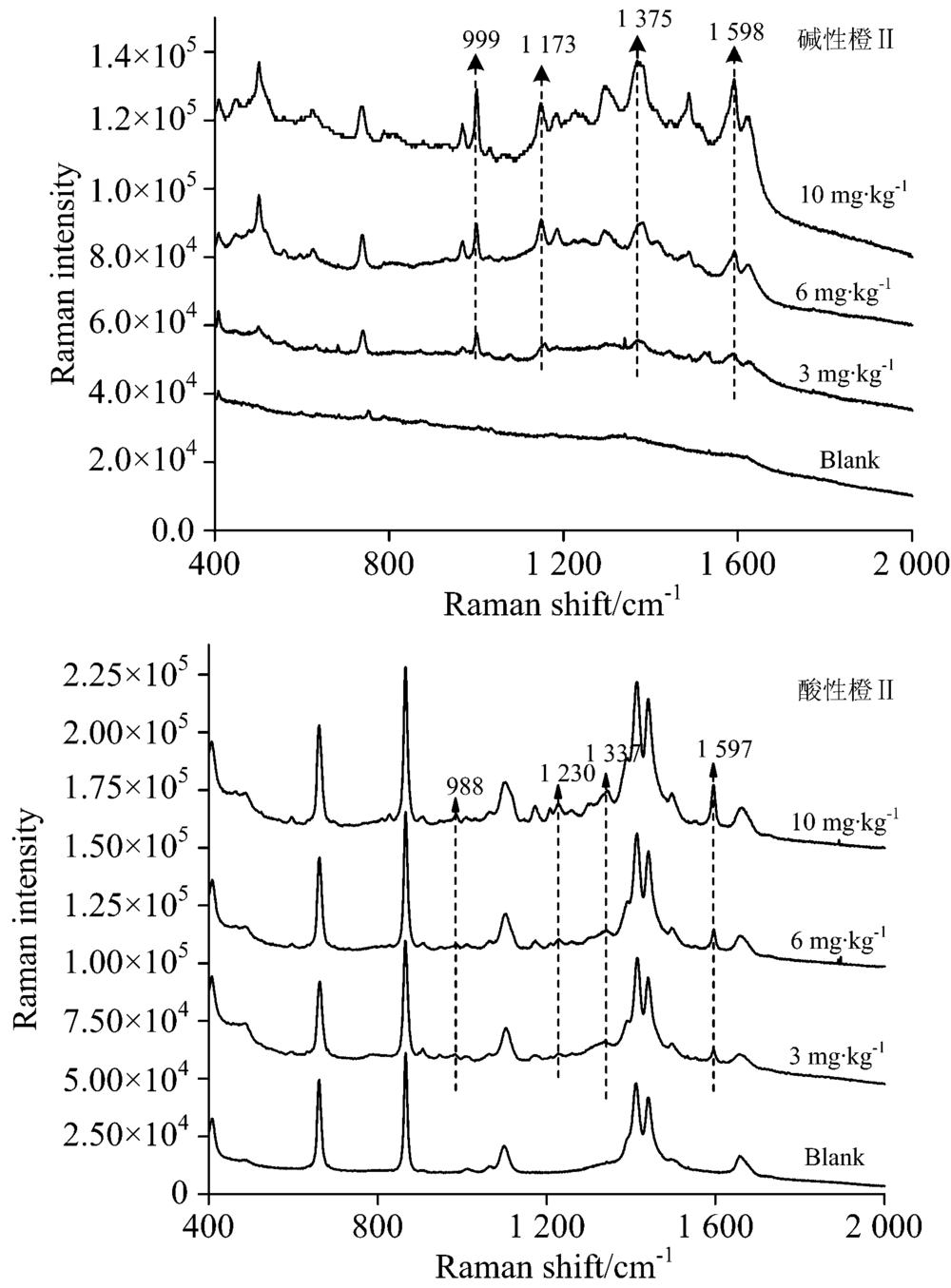 | 图10 腐竹TLC-SERS联用检测光谱Fig.10 SERS of basic orange Ⅱ (a) and acid orange Ⅱ (b) separated from Yuba by TLC |
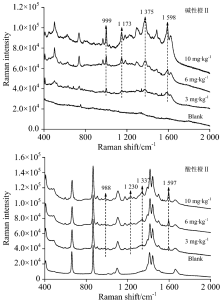
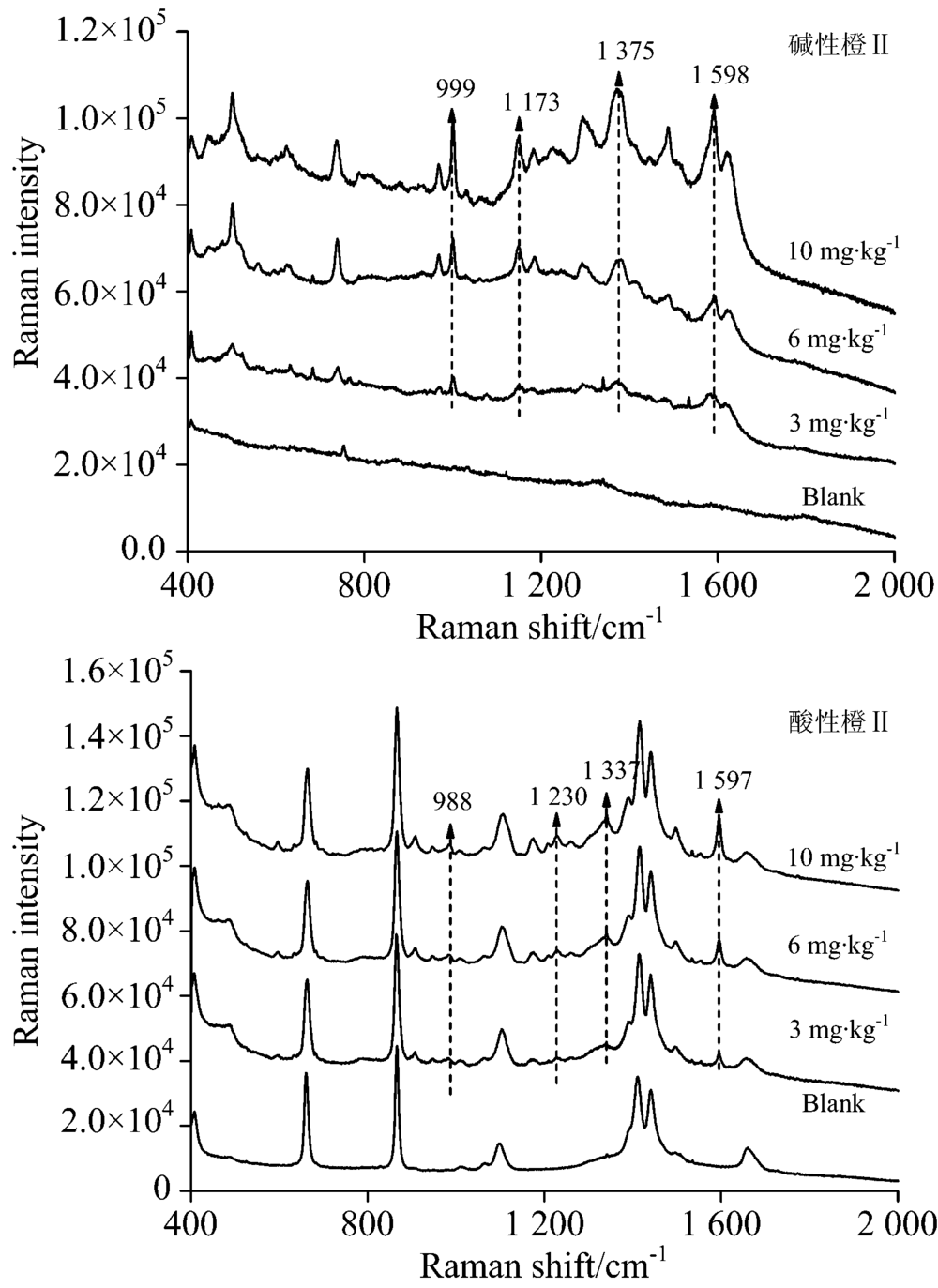 | 图11 油豆皮TLC-SERS联用检测光谱Fig.11 SERS of basic orange Ⅱ (a) and acid orange Ⅱ (b) separated from bean skin by TLC |
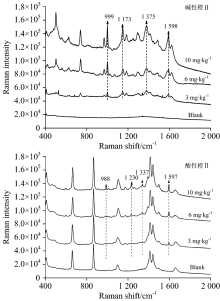
对当地超市购买的腐竹、 油豆皮及卤蛋样品进行高效液相色谱检测, 结果证明均不含有碱性橙Ⅱ 和酸性橙Ⅱ , 默认为空白样品。 然后按1.3.2对三种样品进行三个添加量的添加实验。 图10、 图11、 图12分别是腐竹、 油豆皮、 卤蛋三个添加量及其空白样品经提取后, 以所建立的方法检测获得的拉曼光谱。
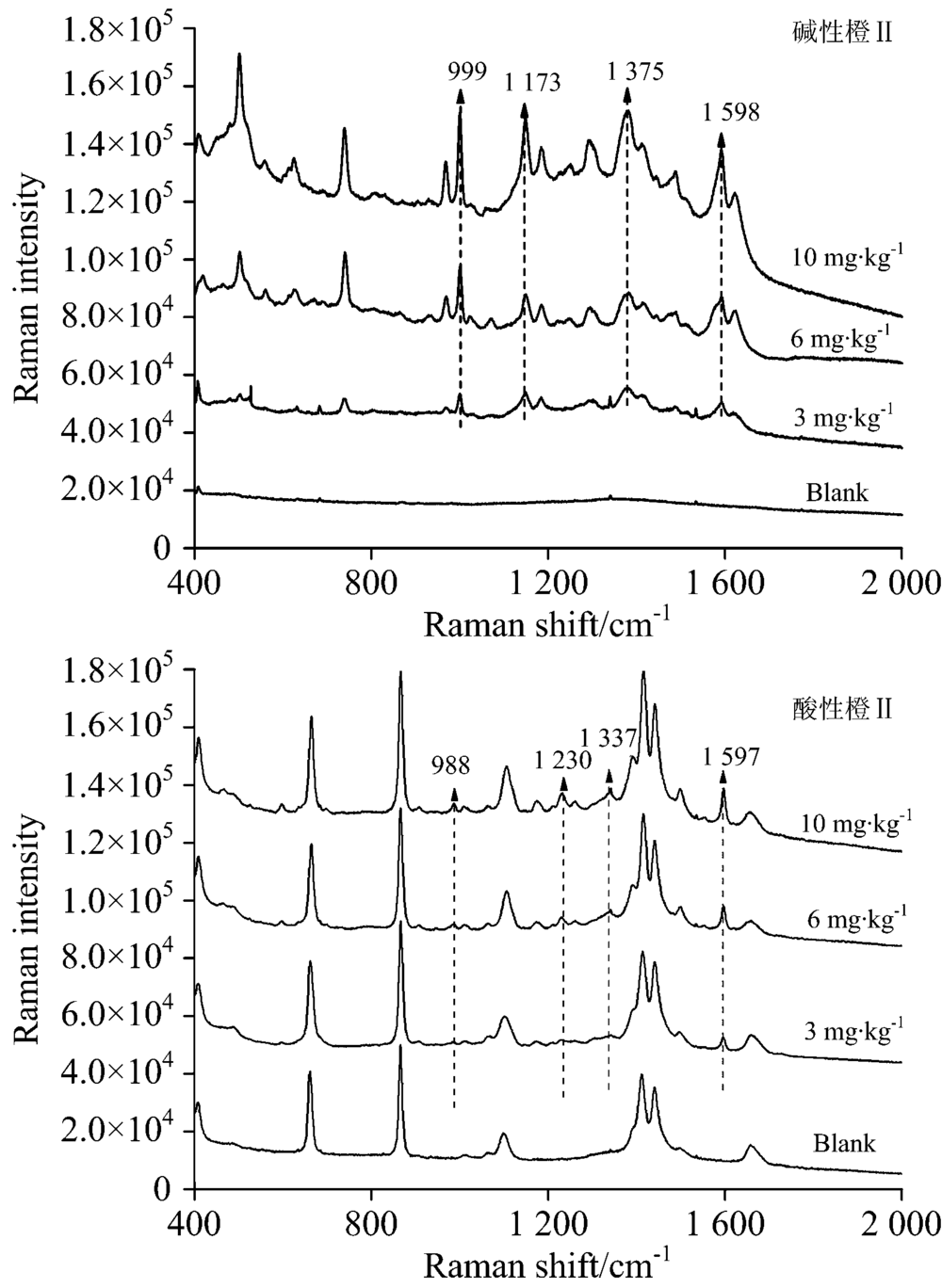 | 图12 卤蛋TLC-SERS联用检测光谱Fig.12 SERS of basic orange Ⅱ (a) and acid orange Ⅱ (b) separated from roasted eggs by TLC |
上述三种食品空白样品检测光谱中没有出现待测物拉曼特征峰, 也没有出现其他未知明显的拉曼峰, 说明提取液中残留的食品复杂基质不会干扰待测物SERS检测。 检测三种食品模拟阳性样品, 获得的高质量的待测物表面增强拉曼光谱, 具有信号强度高, 信噪比高等优点。 碱性橙Ⅱ 四个定性特征峰999, 1 173, 1 375和1 598 cm-1, 酸性橙Ⅱ 四个定性特征峰988, 1 230, 1 337和1 597 cm-1在三个添加量水平内都可以清晰辨别出, 强度也随着添加量的减少逐渐降低, 也未见其他杂峰。 结合薄层板展开图可知薄层色谱法分离了提取液中大部分食品基质, 残余部分也不会干扰待测物SERS检测信号。 由此可见, 所建立的检测方法具有一定实用性。
建立了一种TLC-SERS联用用于快速检测食品中非法添加碱性橙Ⅱ 和酸性橙Ⅱ 方法, 并对实际食品进行添加实验, 用所建立的方法成功检测。 为薄层色谱便携式拉曼光谱在现场食品安全快速检测提供了技术支撑。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|