

{kind=link}
{kind=link}
{kind=link}
{kind=link}
青铜表面钼酸钠缓蚀转化膜XPS深度剖析研究
[范辰骁 , 李昌青, 罗雨佳, 何贝, 杨奔
, 李昌青, 罗雨佳, 何贝, 杨奔* , 金普军* ]
, 李昌青, 罗雨佳, 何贝, 杨奔, 金普军]
|
|


作者简介: 范辰骁, 1998年生,陕西师范大学材料科学与工程学院硕士研究生 e-mail: fanchenxiao2022@163.com
无机钼酸盐缓蚀剂因其绿色、 低毒和高效的特性, 在金属文物防护领域具有广阔的应用前景。 采用不同浓度钼酸钠溶液通过化学沉积法在青铜表面构筑缓蚀转化膜, 并通过电化学测试和XPS深度剖析研究其组成、 结构及性能。 实验结果表明, 青铜试样经1 d浸泡处理后, 采用0.2 mol·L-1钼酸钠溶液处理的试样缓蚀效率较高, 约为50%, 而采用0.02和0.05 mol·L-1钼酸钠溶液处理的试样缓蚀效率相对较低。 随着浸泡时间的延长, 0.2和0.5 mol·L-1钼酸钠溶液处理的试样缓蚀效率逐渐下降, 而0.02和0.05 mol·L-1钼酸钠溶液处理的试样缓蚀效率呈现先上升后下降的趋势。 XPS深度剖析揭示, 缓蚀膜在1.25~5.00 μm范围内发生氧化还原反应, 形成SnO2、 CuO、 Cu2O和MoO2等金属氧化物。 其中Cu元素呈现Cu2O外层、 Cu2O+CuO过渡层和CuO内层的结构, 而Mo元素由表层的$MoO_{4}^{2-}$向内层转变为$MoO_{4}^{2-}+MoO_{2}$, 钼酸根在内层被还原为MoO2。 此外, 试样经钼酸钠溶液处理后出现钼蓝现象, 导致颜色变化明显, 其中0.2 mol·L-1溶液处理3 d的试样色差Δ$E_{Lab}^{*}$约为25, 而0.5 mol·L-1溶液处理3 d的试样色差Δ$E_{Lab}^{*}$约为46。 本研究揭示了钼酸盐缓蚀膜的化学组成及结构特征, 为其在金属文物防护中的应用提供了重要借鉴。
Inorganic molybdate corrosion inhibitors have broad application prospects in conserving metal cultural relics due to their environmentally friendly, low-toxicity, and high-efficiency properties. This study used sodium molybdate solutions of varying concentrations to construct corrosion-inhibiting conversion films on bronze surfaces via chemical deposition. These films' composition, structure, and performance were systematically investigated using electrochemical testing and XPS depth profiling. Experimental results showed that after 1 day of immersion, the bronze sample treated with 0.2 mol·L-1 sodium molybdate solution exhibited a relatively high corrosion inhibition efficiency of approximately 50%. In contrast, samples treated with lower concentrations (0.02 and 0.05 mol·L-1) demonstrated lower efficiencies. With prolonged immersion, the inhibition efficiency of samples treated with 0.2 and 0.5 mol·L-1 solutions gradually decreased, while those treated with 0.02 and 0.05 mol·L-1 solutions initially increased before subsequently decreasing.XPS depth profiling analysis revealed that within the 1.25~5.00 μm depth range, redox reactions occurred, forming metal oxides such as SnO2, CuO, Cu2O, and MoO2. Specifically, Cu exhibited a layered distribution, transitioning from an outer Cu2O layer to an intermediate Cu2O+CuO transition layer and finally to an inner CuO layer. Mo existed predominantly as $MoO_{4}^{2-}$ in the outer film and gradually converted into a mixed form of $MoO_{4}^{2-}$ and MoO2 in the inner layers, with molybdate ions being reduced to MoO2. Additionally, treated samples exhibited a noticeable color change due to the molybdenum blue phenomenon. After 3 days of immersion, the Δ$E_{Lab}^{*}$ color difference of the sample treated with 0.2 mol·L-1 sodium molybdate solution was approximately 25, whereas that of the sample treated with 0.5 mol·L-1 solution was about 46.This study elucidates the chemical composition and structural characteristics of molybdate-based corrosion-inhibiting conversion films on bronze surfaces, providing valuable insights into their application in conserving metal cultural relics.
我国早期文明中先进矿冶和青铜器制作技术与社会文化融合发展, 创建了举世闻名的中国古代青铜文化, 也创造了大批种类丰富、 造型各异和精美绝伦的青铜器。 青铜器保护问题一直受到高度重视, 然而据2003年“ 全国馆藏文物腐蚀损失调查” 结果显示: 全国馆藏文物中50.66%存在不同程度的腐蚀损害, 其中16.5%为重度腐蚀, 而金属文物, 尤其是青铜器, 受损最为严重。
青铜文物长期暴露于土壤埋藏或大气环境中, 受到温湿度变化、 可溶性盐、 臭氧、 腐蚀性气体、 颗粒污染物和光照等多种因素的影响, 导致其主要金属成分(Cu、 Sn和Pb)发生腐蚀[1]。 其中, “ 青铜病” 是青铜器最严重的腐蚀病害之一。 早在19世纪末, 人们就注意到该病害腐蚀迅速且具有传染性[2]。 研究表明Cl-的自催化点蚀是导致青铜器粉状锈形成的主要因素[3]。 馆藏青铜器的腐蚀主要为电化学过程。 潮湿空气在青铜器表面形成薄水膜, 进一步吸收环境中的有害杂质, 形成电解液薄膜, 进而在青铜器表面产生微电池作用, 导致由点及面的电化学腐蚀扩展。 目前针对金属腐蚀的防护措施主要采用表面保护方法, 包括涂装防护、 缓蚀剂防护及电镀层保护等。
涂装防护和缓蚀剂防护法因其高效性和良好的兼容性, 被广泛应用于金属文物防护领域。 然而电镀层保护法会改变文物外观, 因此鲜有应用报道。 目前馆藏青铜器常用的缓蚀剂主要为杂环类有机化合物, 包括苯并三氮唑(BTA)及其衍生物、 2-氨基-5-巯基-1, 3, 4-噻二唑(AMT)及其复配剂、 2-巯基苯并恶唑(MBO)和2-巯基苯并噻唑(MBT)等[4]。 这类有机缓蚀剂不仅能够吸附在青铜金属表面, 还可通过N、 O、 S等杂原子上的孤对电子与铜离子配位, 形成稳定的缓蚀膜[5]。
然而, BTA等杂环类有机缓蚀剂存在毒性及环境不友好性, 并可能因配位反应导致青铜器表面变色, 同时其饱和蒸气压使其稳定性降低[6]。 国内外学者[7]为降低BTA等杂环类有机缓蚀剂的毒性, 陆续研发了咪唑类、 噻唑类和氨基酸等低毒或无毒缓蚀剂, 如南京博物院[8]就围绕AMT缓蚀材料进行了青铜器保护研究。 针对有机缓蚀剂稳定性差问题, 学者们也基于复合成膜和协同缓蚀等思路进行改进, 如: Giuntoli[9]等将聚乳酸与BTA键合作为青铜器的封护材料, 增强膜的抗光老化、 抗湿热老化性能; Giuliani[10]等在壳聚糖中包埋不同的缓蚀剂(如BTA、 MBT)来保护室内青铜器, 聚合物与缓蚀剂的复合提高了涂层的防腐性能和可去除性。
无机缓蚀剂因长效、 高效、 低毒、 环境友好及易去除等特点, 被广泛用于工业缓蚀领域。 无机缓蚀剂通过溶解作用以分子或者离子的形式吸附在金属表面, 或者通过化学反应生成沉淀型转化膜[11], 如: 磷酸氢二钠能够与钢铁表面的铁离子反应, 生成不溶性氧化铁、 磷酸铁和焦磷酸铁等缓蚀转化膜。 在众多的无机缓蚀剂中, 钼酸盐因其具有低毒、 高效和经济等优势, 有望成为工业金属构件防护的普适性缓蚀剂[12]。
四川省文物考古研究所文保中心[13]曾于2000年采用钼酸钠作为缓蚀剂主剂处理了“ 粉状锈” 形成的腐蚀坑, 获得良好的缓蚀效果。 2008年, 胡刚[14]等研究了BTA和Na2MoO4的协同缓蚀作用, 发现青铜表面生成[Cu(Ⅰ )BTA]聚合物保护膜, 并伴随SnO2、 PbO、 MoO3等金属氧化物的沉积, 从而提高膜层的致密性和耐腐蚀性。 2017年Zhou[15]等进一步发现BTA和Na2MoO4复合缓蚀剂同样适用于钢材缓蚀, 可促进锈蚀产物FeOOH向稳定的Fe2O3转化。
尽管关于钼酸盐在金属缓蚀机制方面的研究相对较少, 但现有研究成果已揭示了一些重要问题, 例如: Bentley[16]等发现
综上所述, 钼酸盐可在青铜器表面吸附成膜, 抑制氯离子腐蚀, 在青铜文物缓蚀防护中发挥了重要作用。 为深入探究其对青铜表面的作用机制, 本文采用化学溶液沉积法构筑青铜试样表面缓蚀转化膜, 通过极化曲线(Tafel)、 交流阻抗技术(EIS)、 扫描电镜(SEM)、 X射线光电子能谱(XPS)和色差分析(CIELAB)等技术研究缓蚀膜的组成、 结构和性能, 进一步揭示钼酸盐在青铜表面形成缓蚀转化膜的化学组成和结构特征, 为青铜文物和铜基合金材料保护提供理论基础与技术支撑。
青铜试样购置于义鑫铜材公司, 为3.0 mm厚度的C5191模拟青铜片(成分见表1)。 青铜片剪切为1 cm× 1 cm的方片作为模拟样片, 并依次采用400, 800, 1 200, 1 500和2 000#的金相砂纸打磨至镜面, 随后将样片依次在丙酮和乙醇溶液中分别超声清洗10 min, 以去除表面油脂, 最终置于干燥器中备用。
| 表1 C5191青铜合金的化学成分(wt%) Table 1 Chemical compositions of C5191 bronze alloys (wt%) |
准确称量钼酸钠粉末0.484、 1.210、 4.839和12.097 g, 分别溶解于100 mL去离子水(置于100 mL烧杯中), 经磁力搅拌配制成0.02、 0.05、 0.2和0.5 mol· L-1的钼酸钠溶液。 随后将预处理后的青铜试样悬挂浸泡于上述溶液中, 并在50 ℃水浴及300 r· min-1搅拌条件下进行反应。 分别在1、 2和3 d后取出试样, 自然干燥后进行表征分析。
采用CHI660E型电化学工作站进行电化学测试, 其中工作电极为浸泡处理后的模拟青铜片, 参比电极为217型饱和甘汞电极(SCE), 辅助电极为铂片电极, 电解质溶液为3.5%(质量分数)NaCl水溶液, 所有实验均在室温下进行。 测试前电极需预先浸泡于电解液中直至开路电位稳定。 电化学阻抗谱测试(EIS)在开路下进行, 采用正弦交流信号, 振幅为10 mV, 频率范围为104~10-2 Hz, 采用Zview软件对Nyquist图谱进行拟合。 Tafel极化曲线扫描电位范围为-0.4~0.6 V, 扫描速率为0.01 V· s-1。
采用日本HITACHI日立公司生产的SU3500钨灯丝扫描电子显微镜在10 kV电压下观察样品表面形貌。
采用赛默飞世尔科技有限公司生产的Escalab Xi+型X射线光电子能谱仪对样品表面进行元素价态分析, 并通过深度剖析功能探究缓蚀转化膜不同深度下元素的化学状态。
采用广东三恩时智能科技有限公司生产的SC-10型便携式色差仪对缓蚀转化膜的色度进行分析, 侦测器为硅光电二极管阵列, 精度Δ
极化电位通常用于衡量金属的腐蚀难易程度, 而极化电流密度则反映腐蚀速率[20]。 图1为青铜试样在0.02、 0.05、 0.2和0.5 mol· L-1钼酸钠溶液中浸泡1、 2和3 d后, 在3.5% NaCl水溶液腐蚀介质中测试的Tafel极化曲线图。 从图中可以看出, 随着浸泡时间的延长, 极化电位整体呈负移趋势, 其中0.02和0.05 mol· L-1 低浓度钼酸钠溶液处理的试样负移幅度较大, 而0.2和0.5 mol· L-1 高浓度溶液处理的试样极化电位变化较小。 这表明低浓度钼酸盐处理的试样随着浸泡时间增加, 缓蚀膜缓蚀效率明显降低。
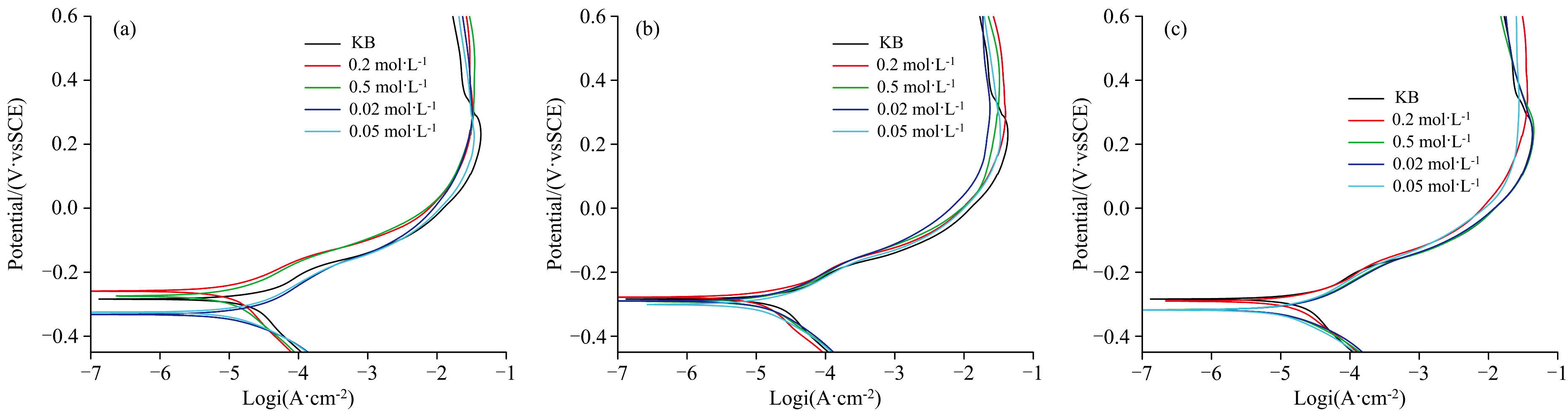 | 图1 模拟青铜浸泡不同浓度钼酸钠水溶液后的极化曲线图 浸泡时间(a): 1 d; (b): 2 d; (c): 3 dFig.1 Polarization curves of simulated bronze immersed in different concentrations of sodium molybdate aqueous solution Immersion time (a): 1 day; (b): 2 days; (c): 3 days |
表2列出了空白样品及不同浸泡时间样品的电化学参数, 结果表明青铜试样在0.2和0.5 mol· L-1 钼酸钠溶液中浸泡1 d后, 缓蚀效率达到约50%, 随后随浸泡时间延长有所下降, 但整体保持较高水平。 相比0.02和0.05 mol· L-1低浓度钼酸钠溶液缓蚀效率, 青铜试样经过高浓度溶液处理形成钼酸盐缓蚀膜的缓蚀效率较高。
| 表2 钼酸钠处理后的模拟青铜在3.5%NaCl溶液中的极化曲线参数及缓蚀效率 Table 2 Polarization parameters and corrosion inhibition efficiency of sodium molybdate-treated simulated bronze in 3.5% NaCl solution |
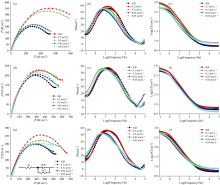
钼酸钠溶液处理后的模拟青铜试样的腐蚀行为与其在腐蚀介质中的传质特性密切相关。 为了进一步探究其腐蚀过程, 进行了电化学阻抗谱(EIS)测试, 采用ZView软件通过等效电路模型来拟合EIS曲线(图2), 其中Rs为溶液电阻, Rct为电荷转移电阻, CPE代表适应电容回路的恒定相位元件, Rct值通常越大表明耐腐蚀性能越好[21]。
 | 图2 模拟青铜在钼酸钠水溶液中浸泡不同时间后的交流阻抗谱 (a), (d), (g)为1, 2和3 d的Nyquist图; (b), (e), (h)为1, 2, 3 d的Bode相位角图; (c), (f), (i)为1, 2, 3 d的Bode阻抗图Fig.2 Electrochemical impedance spectroscopy (EIS) of simulated bronze samples after different immersion times in sodium molybdate solution (a), (d), (g) are Nyquist plots for 1 day, 2 days, and 3 days; (b), (e), (h) are Bode phase angle plots for 1 day, 2 days, and 3 days; (c), (f), (i) are Bode impedance plots for 1 day, 2 days, and 3 days |
图2为模拟青铜试样在不同浓度钼酸钠溶液中浸泡不同时间后的Nyquist图和Bode图, 其中Nyquist曲线[图2(a、 d、 g)]表明所有样品均呈现相似的容抗弧形状, 但其半径存在差异。 容抗弧与腐蚀过程中的电荷传递控制有关, 半圆形容抗弧代表均匀腐蚀过程, 而容抗弧形状畸变意味着腐蚀行为不完全均匀, 存在局部腐蚀现象[22]。
钼酸钠溶液处理后的试样在整个频率范围内表现出比未处理试样更高的阻抗值, 如: 经0.2 mol· L-1钼酸钠溶液处理 1 d 后, 青铜试样呈献圆形且较大的容抗弧, 其电荷转移电阻Rct值为1 049 Ω , 显著高于空白样品的605.8 Ω 。 因此较大的容抗弧半径与较高的电荷转移电阻表明形成的钼酸盐缓蚀转化膜分布均匀, 能够有效阻挡腐蚀介质渗透, 从而降低腐蚀速率。 然而随着浸泡时间的延长, 容抗弧半径逐渐缩小, Rct值同步降低, 反映出缓蚀膜的耐腐蚀性能有所下降。
根据图2(b、 e、 h)的Bode相位角图, 钼酸钠溶液处理后的模拟青铜试样在整个频率范围内仅出现一个时间常数, 这通常对应于电荷转移控制的腐蚀过程[23]。 图中相位角随频率呈抛物线分布, 表现出典型的容抗行为, 所有样品的最大相位角均在50° ~60° 之间。 一般而言, 相位角增大表明腐蚀速率降低。 其中经0.2 mol· L-1钼酸钠溶液浸泡1 d的试样相位角最高, 表现出最佳缓蚀效率。 相比之下, 0.02和0.05 mol· L-1低浓度钼酸钠溶液处理的试样在短时间内相位角有所增加, 但随浸泡时间延长逐渐下降, 进一步验证了低浓度钼酸盐形成缓蚀膜的稳定性较差。
图2(c、 f、 i)的Bode阻抗图表明钼酸钠溶液处理前后的青铜试样均表现出相似的容抗弧形状。 此外经钼酸钠溶液处理的模拟青铜在低频区的|Z|值明显高于未处理试样, 表明其耐腐蚀性能得到了不同程度的提升[24]。
图3为模拟试样在0.2和0.5 mol· L-1钼酸钠溶液中浸泡1和3 d后的SEM形貌图, 结果表明青铜试样经0.2 mol· L-1钼酸钠溶液浸泡3 d后, 在其表面形成较为厚实且均匀的沉积层; 而在0.5 mol· L-1钼酸钠溶液中浸泡3 d后则出现了大颗粒的树枝状结晶形貌。
 | 图3 模拟青铜试样经钼酸钠水溶液浸泡1 d [(a) 0.2 mol· L-1; (c): 0.5 mol· L-1)和3 d后[(b): 0.2 mol· L-1; (d): 0.5 mol· L-1)扫描电子显微照片Fig.3 SEM images of treated bronze surface after 1 day [(a) 0.2 mol· L-1; (c): 0.5 mol· L-1) and 3 days [(b): 0.2 mol· L-1; (d): 0.5 mol· L-1) immersion in aqueous sodium molybdate solutions |
根据Ostwald熟化理论[25], 大颗粒晶粒因其内部分子间的紧密连接, 能量通常低于小颗粒晶粒。 在长时间浸泡过程中, 小颗粒晶粒易发生迁移和团聚, 最终形成能量较低的大颗粒晶粒, 从而促使缓蚀膜呈现出大颗粒堆积状, 进而导致试样耐腐蚀性能的降低。 Ostwald熟化理论合理地解释了经1 d浸泡处理的试样缓蚀效率优于浸泡3 d的原因。 此外, Essom[26]的研究合理解释了本实验中随钼酸钠溶液浓度增加, 缓蚀效率表现出先升高后降低的现象, 即高浓度条件下颗粒更易发生快速聚集, 从而形成疏松结构降低膜层的致密性和缓蚀效率。
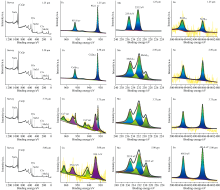
为进一步解析钼酸钠缓蚀膜的组成结构, 本研究采用XPS深度剖析方法, 研究了0.5 mol· L-1钼酸钠溶液浸泡3 d 后形成的缓蚀转化膜在1.25~5.00 μm深度范围内的Cu、 Mo和Sn元素的化学状态。 XPS检测元素浓度以原子分数(at%)表示, 并经过归一化计算, 以确定各元素在不同刻蚀深度的相对含量。 XPS深度剖析检测深度为1.25、 2.50、 3.75和5.00 μm, 并采用Avantage软件对数据进行校正、 分峰拟合及元素含量计算, 结果如图4和表3所示。
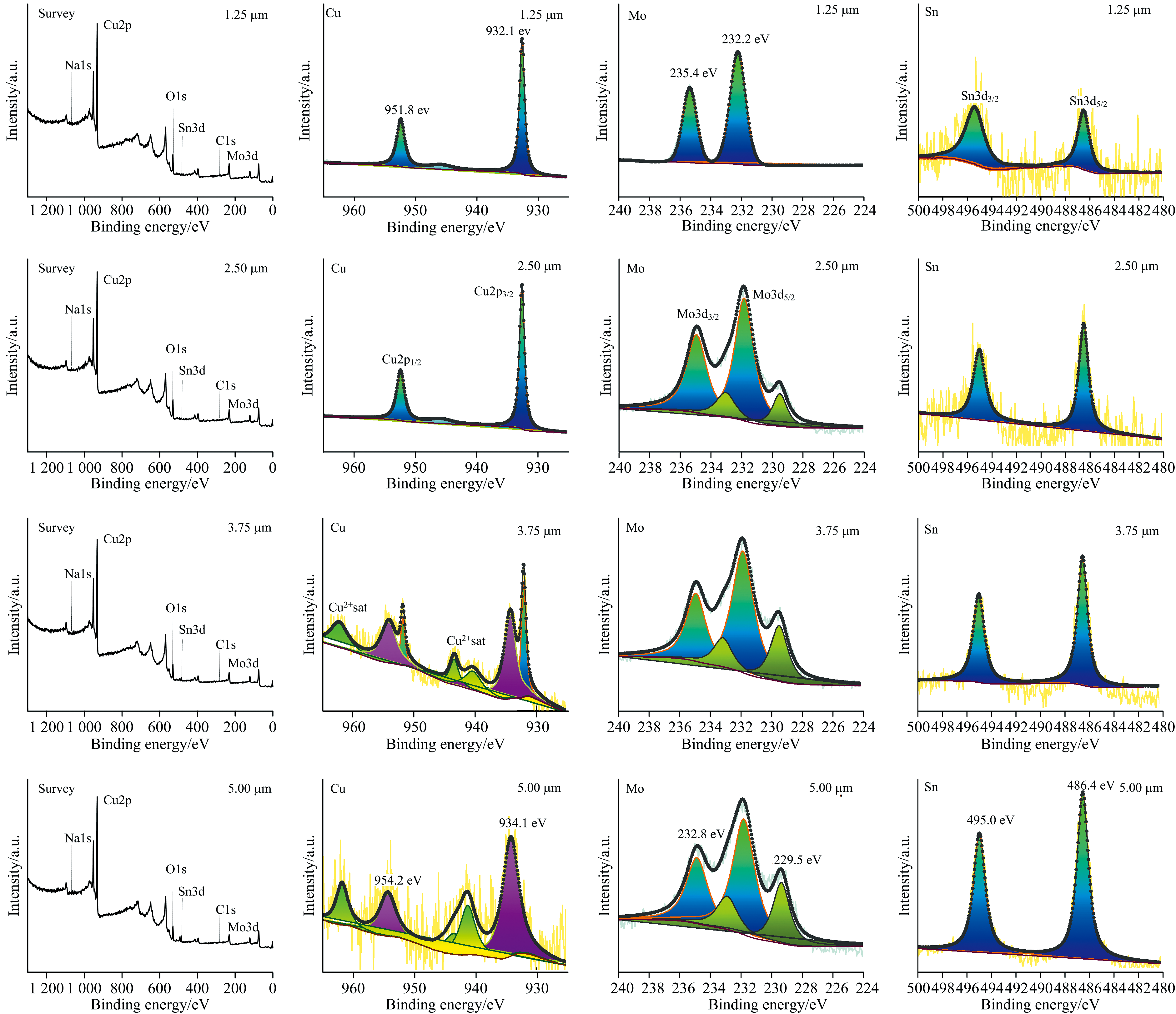 | 图4 缓蚀转化膜中1.25~5.00 μm的XPS衍射图谱深度剖析图Fig.4 XPS depth profile analysis of the conversion film from 1.25 to 5.00 μm |
| 表3 不同刻蚀深度下元素原子含量数据表(at%) Table 3 Elemental content attained by XPS depth profile analysis at different etching depths (at%) |
Cu元素的XPS深度剖析结果表明: 在1.25 μm深度处观察到932.1和951.8 eV两个明显的峰, 并伴有弱卫星峰, 可归属于Cu2O[27]; 在3.75 μm深度处观察到了Cu2O特征峰, 以及位于934.1和954.2 eV处的CuO特征峰[28]; 值得注意的是, 在5.00 μm深度处仅观察到位于934.1和954.2 eV处的CuO特征峰, 未检测到Cu2O特征峰。 综上所述, 钼酸钠转化膜中的Cu元素化学状态呈现出分层特点: 表层为Cu2O, 中间层为Cu2O+CuO的过渡层, 内层为CuO。
Mo元素XPS深度剖析表明: 在1.25 μm深度观察到位于232.2和235.4 eV处的
Sn元素XPS深度剖析表明: 在486.4和495.0 eV处出现了SnO2的特征峰, 表明青铜中的锡发生了氧化反应。 锡元素在缓蚀转化膜中原子含量从外层1.25 μm深度位置的0.32 at%逐渐增加到内层5.00 μm处的1.74 at%。 Cu元素在缓蚀转化膜中原子含量则高达50 at%左右, 说明铜和钼氧化物是缓蚀膜中主要氧化物。
综上所述, XPS深度剖析实验反映钼酸盐在沉积吸附过程中会与金属发生氧化还原反应被还原为钼的氧化物, 而青铜中Cu和Sn被氧化形成Cu2O、 CuO和SnO2。 这些金属氧化物生成有助于增强缓蚀转化膜的附着力、 致密性和稳定性, 从而提高膜层的耐腐蚀性能。
色差分析可以反映试样前后颜色的变化程度, 对于评估保护材料的效果至关重要, 一般依据CIELAB公式计算得出色差值, 如
其中, Δ
采用三恩时SC-10型便携式色差仪对模拟试样进行色度分析, 并以未处理试样的色度数据作为基准。 测试结果表明经0.2 mol· L-1钼酸钠溶液浸泡3 d的试样, 其色差Δ
钼酸盐在金属表面处理过程中易形成钼蓝现象, 系钼基氧化物导致金属表面颜色发生了变化。 “ 整旧如旧” 及“ 不改变文物原状” 是文物保护基本原则, 要求文物在保护处理前后保持色泽基本不变。 一般金属文物表面都附着一层绿色锈蚀, 成为吸附和隔离钼酸盐的重要缓冲区域, 在采用钼酸盐作为缓蚀剂时需要考虑其用量和稳定钼酸盐的辅助措施。
(1)钼酸盐可通过化学溶液沉积法在青铜试样表面形成缓蚀转化膜, 研究表明试样经0.02, 0.05, 0.2和0.5 mol· L-1 钼酸钠溶液缓蚀处理青铜样品, 在3.5% NaCl溶液中缓蚀效率存在明显变化规律, 即0.2 mol· L-1钼酸钠溶液浸泡1 d的试样缓蚀效率最高约为50%。 随着浸泡时间延长, 0.2和0.5 mol· L-1较高浓度处理的试样缓蚀效率逐渐下降, 而0.02和0.05 mol· L-1较低浓度处理的试样缓蚀效率则呈现先上升后下降的趋势。
(2)缓蚀膜中存在SnO2, CuO, Cu2O和MoO2等金属氧化物, 表明钼酸盐在吸附沉积过程中发生了氧化还原反应。 XPS深度剖析显示Sn和Cu元素的含量在缓蚀转化膜中由内向外逐渐降低, 其中Sn在缓蚀膜中的迁移程度低于Cu, 而Cu元素由外向内依次呈现Cu2O、 CuO+Cu2O和CuO的氧化物结构。 同时, Mo元素在缓蚀膜表层主要以
(3)色差分析显示钼酸钠溶液处理后的青铜试样颜色变化明显, 经0.2 mol· L-1钼酸钠溶液浸泡3 d的样品色差Δ
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|