


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
苯甲酸和山梨酸混合物的太赫兹吸收峰形成机理解析
[李文文 , 燕芳
, 燕芳* , 刘洋硕]
, 燕芳, 刘洋硕]
|
|


作者简介: 李文文,女, 2000年生,内蒙古科技大学自动化与电气工程学院硕士研究生 e-mail: 19801524408@163.com
以苯甲酸和山梨酸为研究对象, 采用太赫兹时域光谱系统(THz-TDS)分别测得二者的单质与混合样片在0.5~2.2 THz的实验光谱, 将光谱数据进行预处理后获得两种物质的太赫兹(THz)吸收光谱, 根据吸收光谱中特征吸收峰峰位可以精准实现对上述两种物质的定性鉴别。 利用Genmer组件自行构建了苯甲酸和山梨酸的混合物二聚体团簇构型, 结合密度泛函理论(DFT)对其单分子、 晶胞及混合物团簇的构型进行了结构优化、 频率计算和筛选。 采用势能分布分析了特征吸收峰的振动模式, 获取模拟光谱后结合基于Hirshfeld原子空间划分的独立梯度模型(IGMH)和相互作用区域指示函数(IRI)两种相互作用可视化分析方法针对混合样片中异常吸收峰的振动模式进行识别, 并分析了分子内化学键和分子间相互作用的可视化特征。 研究发现, 基于混合物团簇构型的模拟谱中的特殊吸收峰是由于分子之间的氢键振动引起的, 进一步说明了苯甲酸和山梨酸及其混合物在THz频段的特征吸收峰主要源于分子间氢键作用和分子内化学键作用引起的集体振动模式。 通过混合体系的团簇构型构建及筛选深入解析了混合物太赫兹吸收谱中特征吸收峰的成因, 重点探讨了分子内外相互作用、 氢键效应等因素对吸收峰的影响, 为晶体结构的表征拓展了一种新的方法, 显著提升了混合体系吸收光谱在谱图解析中的准确性和可靠性, 为后续进一步基于光谱数据的定量分析提供了保障。
This study focuses on benzoic acid and sorbic acid, using a terahertz time-domain spectroscopy (Terahertz Time-Domain Spectroscopy, THz-TDS) system to measure the experimental spectra of their pure substances and mixed pellets in the 0.5~2.2 THz range.After preprocessing the spectral data, the two substances' terahertz (Terahertz, THz) absorption spectra were obtained. The qualitative identification of the above two substances can be accurately achieved based on the peak positions of the characteristic absorption peaks in the absorption spectra. In this paper, the dimer cluster configurations of the benzoic acid and sorbic acid mixture were constructed using the Genmer component. Structural optimizations, frequency calculations, and screening were conducted for their single molecules, unit cells, and mixture clusters using density functional theory (DFT). The potential energy distribution was used to analyze the vibration modes of the characteristic absorption peaks. Combining the simulated spectra with two interaction visualization methods—Independent Gradient Model based on Hirshfeld atom partitioning (IGMH) and the Interaction Region Indicator (IRI)—the vibration modes of abnormal absorption peaks in the mixed pellet were identified. The molecular and intermolecular interaction features were also visualized. The study found that the special absorption peaks in the simulated spectra of the mixture cluster configuration were caused by hydrogen bond vibrations between molecules. This further illustrates that the characteristic absorption peaks of benzoic acid, sorbic acid, and their mixtures in the terahertz frequency range mainly originate from collective vibration modes induced by intermolecular hydrogen bonds and intramolecular chemical bonds. By constructing and screening cluster configurations of mixed systems, this work deeply investigates the causes of characteristic absorption peaks in the terahertz absorption spectra of mixtures. It highlights the impact of intramolecular and intermolecular interactions and hydrogen bond effects on absorption peaks. This study introduces a novel method for characterizing crystal structures, significantly enhancing the accuracy and reliability of spectral interpretation for mixed systems. It also lays a foundation for subsequent quantitative analyses based on spectral data.
太赫兹(Terahertz, THz)波是频率范围在0.1~10 THz的电磁波, 广泛应用于分子振动和转动模式的研究。 由于太赫兹频段能够覆盖极性分子和生物大分子的特征吸收区域, 其在无损检测领域具有巨大潜力[1]。 苯甲酸和山梨酸分子结构中的氢键等相互作用和晶体振动模式在太赫兹频段内均表现出显著的特征吸收峰, 能够为相关物质的分子光谱学特性分析提供关键数据支持[2, 3, 4]。 目前已有研究大多集中于单一分子的特征吸收峰机理解析, 而针对混合体系的团簇构型构建与特征吸收峰成因分析仍显不足。
近年来, 针对苯甲酸和山梨酸物质的太赫兹光谱特性及振动模式已有一些研究成果。 2013年郑转平等[5]基于太赫兹时域光谱(Terahertz time-domain spectroscopy, THz-TDS)技术测得了苯甲酸及苯甲酸钠的特征吸收谱, 归属了苯甲酸的实验特征吸收峰, 并结合密度泛函理论对苯甲酸钠的单分子结构进行了优化和振动模式的计算。 研究发现, 苯甲酸钠的离子键显著影响了分子内共价键的键长与键角, 以及晶胞内分子间的排列, 导致其与苯甲酸的吸收谱存在差异。 此文中主要集中于单一物质的单分子结构光谱特性分析, 尚未针对该物质其他分子结构, 如晶胞等进行特征吸收峰的机理解析; 2021年, 李天莹等[6]对苯甲酸、 山梨酸和木糖醇这三种富含氢键的典型食品添加剂进行了太赫兹光谱特性的实验测量与理论分析, 文中利用CRYSTAL14软件包计算了周期性晶体结构, 通过在传统密度泛函理论模型中引入色散校正, 提高了模拟光谱的预测精度, 并提出了对单质晶胞振动模式的理论解释。 研究指出, 经过色散校正的模型能够更精确地描述富含氢键的体系特征, 色散校正在准确描述分子间弱相互作用(如氢键和范德华力)方面具有重要意义。 这些研究工作为食品添加剂分子的光谱特性解析提供了重要理论及方法参考, 但文中主要针对单质体系, 且重点在提高模拟光谱的预测精度, 未能进一步探索复杂混合体系的吸收峰规律及形成机制。 综上所述, 前期的研究大多集中于针对单质的结构进行振动分析, 而涉及混合物体系的光谱行为及其异常吸收现象的解析研究较少, 对混合物体系特征吸收峰的形成机制亦缺乏深入探讨。 虽然前人工作中已采用过混合物团簇构建和量子化学模拟方法, 使用独立梯度模型(independent gradient model, IGM)和相互作用区域指示函数(interaction region indicator, IRI)方法对分子间相互作用进行了可视化分析, 并成功解释了特殊吸收峰的成因。 但这些研究多集中于氨基酸等特定小分子, 尚未覆盖其他种类, 研究对象具有一定局限性, 且鲜见针对两种或两种以上混合物吸收谱中的特殊吸收现象进行解析的相关研究。
本文以苯甲酸和山梨酸为研究对象, 针对二者混合样片中的吸收峰偏移与缺失的异常吸收现象进行研究。 本文参考了前人关于混合物的团簇构建方法, 构建了苯甲酸和山梨酸的混合物二聚体团簇模型, 结合密度泛函理论分别对苯甲酸和山梨酸的单分子、 晶胞及混合物团簇进行了优化与振动模式计算, 并利用势能分析(potential energy distribution, PED)匹配了振动模式, 利用基于Hirshfeld原子空间划分的独立梯度模型(independent gradient model based on hirshfeld partition, IGMH)、 IRI方法对其分子间的相互作用进行了可视化分析, 由此解析了混合物体系中异常吸收峰的振动模式及其成因。 研究结果表明, 混合物团簇中的特定吸收峰主要来源于分子间氢键作用及分子内化学键的集体振动模式。 本研究不仅丰富了食品添加剂太赫兹光谱的理论基础, 还提出了一种基于团簇建模的混合物体系分析策略, 为复杂体系的晶体结构表征及光谱信息提取提供了重要的理论与技术支持。
采用北京市工业波谱成像工程技术研究中心的TPS1000D透射式太赫兹时域光谱系统进行实验光谱数据采集, 实验装置与设备如文献[7]所述。
实验用样品苯甲酸、 山梨酸均采购自阿拉丁试剂网, 聚乙烯(Polyethylene, PE)采购于西格玛奥德里奇(Sigma-Aldridge)公司。 首先将所用样品(苯甲酸与山梨酸)与聚乙烯按实验设定的质量比称量好并倒入玛瑙研钵中充分研磨, 然后用压片机以5 MPa的压力压制5 min, 制为厚度1.35~1.40 mm左右, 直径为13 mm的圆形薄片。 分别制作浓度梯度为10%的苯甲酸与山梨酸单质样片各3个, 以及苯甲酸和山梨酸的混合样品3个, 样片制备信息表如表1。
| 表1 样品制备信息表 Table 1 Sample preparation information sheet |
实验时, 为降低样片颗粒度及均匀度所带来的误差, 对每个样片的同一点位均进行三次测量, 并取其平均值保存为最终结果, 3组9个样品共获得27组光谱数据。
经过太赫兹时域光谱系统获得样片的时域信号后, 根据Dorney等和Duvillaret等提出的光学参数提取模型, 结合快速傅里叶变换(fast Fourier transform, FFT)计算样片的折射率与吸收系数, 如式(1)和式(2)所示[8, 9]。
式(1)中, c为光速, d为样片厚度, ω 为时域信号经傅里叶变换(FFT)后的角频率, φ (ω )为样品信号与参考信号之间的相位差。
式(2)中, A(ω )为吸收系数, Esam(ω )为样品信号的幅频特性, Eref(ω )为参考信号的幅频特性, d为样片厚度, ns(ω )为试样的折射系数。
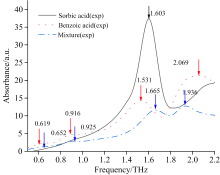
图1为苯甲酸和山梨酸在0.5~2.2 THz波段的特征吸收谱。 首先, 如图可知苯甲酸共有4个特征吸收峰, 分别位于0.619、 0.916、 1.531和2.069 THz, 而山梨酸则仅在1.603 THz有1个较明显的特征吸收峰, 故可根据特征吸收峰的峰位及峰数实现上述两种添加剂的定性鉴别。 其次, 图1中混合物(苯甲酸和山梨酸)样片的吸收光谱中属于苯甲酸的1.531 THz处的单质吸收峰在吸收谱中缺失了, 属于特殊吸收现象, 这将影响混合样片通过吸收峰进行定性识别的准确性, 进一步对后续的精确定量分析形成干扰, 故本文结合量子化学分析方法对上述特殊吸收现象进行了进一步的分析。
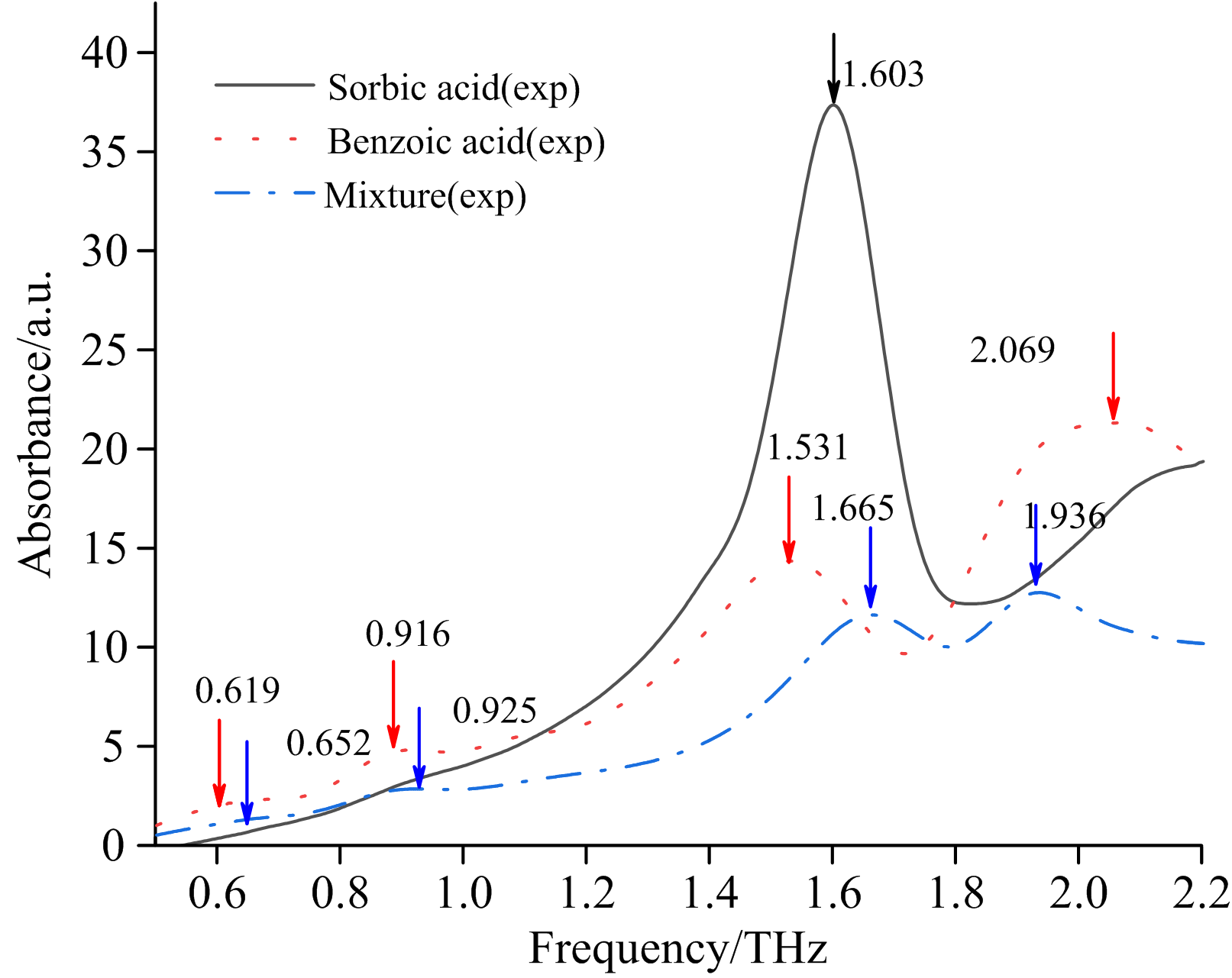 | 图1 苯甲酸、 山梨酸、 苯甲酸和山梨酸混合物的太赫兹实验谱Fig.1 Benzoic acid, sorbic acid, and benzoic acid-sorbic acid mixture terahertz experimental spectra |
首先, 从剑桥晶体数据库中心(Cambridge Crystallographic Data Centre, CCDC)提取苯甲酸和山梨酸的单分子及晶胞构型, 并利用Gaussian 09W展开理论模拟谱的计算。 结果表明, 二者单分子构型的吸收模拟谱与实验谱的特征吸收峰不符, 这是由于单分子构型的结构过于单一, 无法充分体现其在自然条件下的微观状态[10]。 相比之下, 晶胞构型中包含多个单分子构型的排列, 更全方位地分析思考了分子内化学键、 分子间的氢键及范德华作用的影响, 能更准确地模拟分子间和分子内的相互作用, 所以采用晶胞构型进行模拟谱的计算获取。
其次, 采用苯甲酸与山梨酸的晶胞构型, 基于杂化泛函的B3LYP(Becke, 3-parameter, Lee-Yang-Parr)[11]结合Grimme的DFT-D3(BJ)[12]色散矫正和基组6-311g对两种物质结构进行优化和频率的计算, 获得了它们的太赫兹理论模拟吸收谱, 见图2。 由图2(a)中可以看出苯甲酸的模拟谱和实验谱中吸收峰个数相同, 峰位相似。 由图2(b)可知, 山梨酸的模拟谱与实验谱基本吻合, 均有1个极为显著的吸收峰, 且峰位相似。 这表明本文选择的晶胞构型和计算基组是合适的。 而峰位上存在的偏差是由于理论模拟谱是在绝对零度(0 K)下计算获取的, 但实验谱是在常温(298 K)下测量得到的, 随着温度的升高, 热膨胀效应使两个原子之间的键长增加, 分子间距离也随之增大, 导致晶胞的晶格参数(如晶胞体积)发生改变。 随着晶胞体积的增加, 分子间相互作用的力度会降低, 导致实验光谱与模拟光谱在吸收峰峰位上出现偏移[13]。
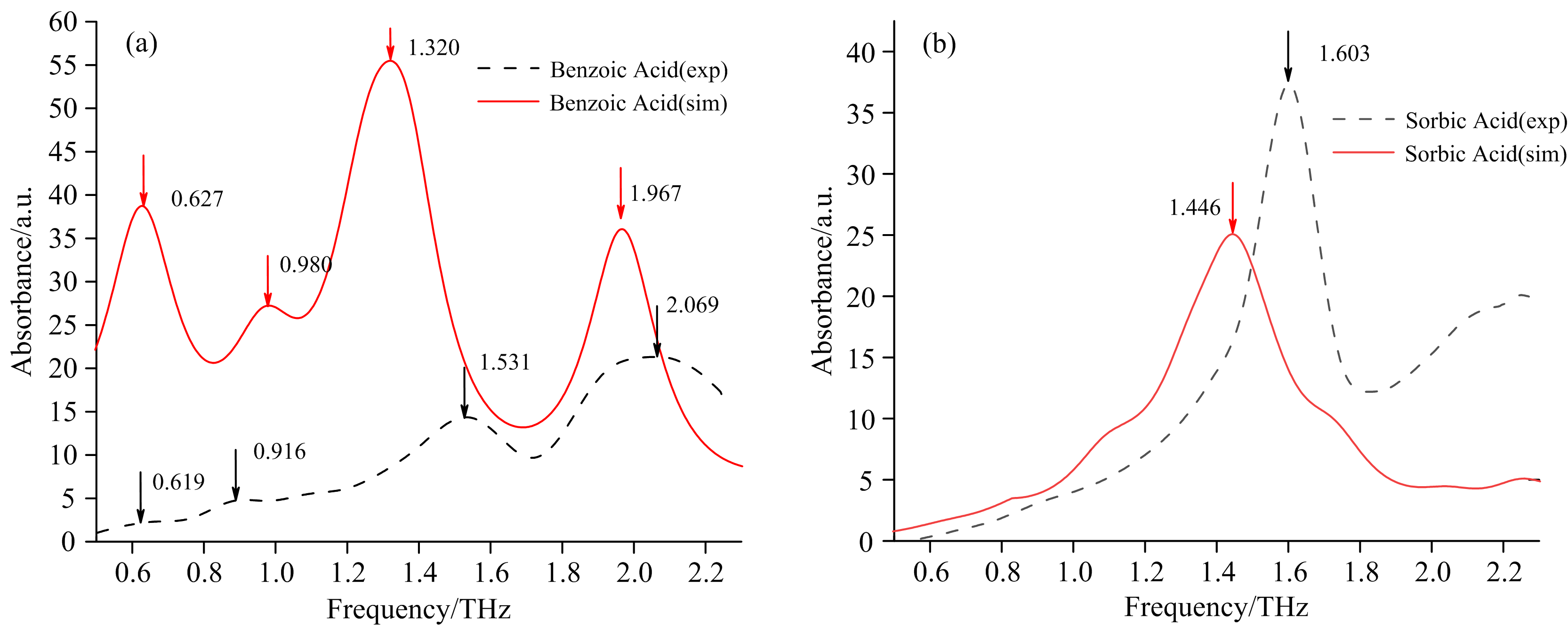 | 图2 苯甲酸(a)和山梨酸(b)实验谱、 晶胞构型模拟谱对比Fig.2 Comparison of experimental spectra and simulated spectra for benzoic acid (a) and sorbic acid (b) |
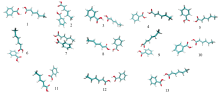
为进一步研究苯甲酸与山梨酸混合物的吸收峰形成机理, 本文进行了混合物的模拟谱计算。 由于CCDC数据库中没有苯甲酸和山梨酸混合物的晶胞构型, 故本文利用Molclus中的Genmer[14]组件构建了500个期望初始团簇构型。 以苯甲酸为团簇中心, 加入山梨酸单质, 通过计算获得了含有500个初始构型的能量值和分子结构坐标信息的xyz文件, 但在这些初始团簇构型中存在一些结构相似, 且不合理的构型, 故先用专门针对原子之间的弱相互作用的半经验方法PM6-DH+在MOPAC[15]下进行了预优化, 优化完成后按结构能量由低到高排序, 筛选出124个团簇构型。 在此基础上再用密度泛函理论(density functional theory, DFT)-B3LYP-6-311g* * 结合D3矫正的方法进行结构优化计算, 筛选得到50个苯甲酸和山梨酸混合物的团簇构型。 最后基于结构的稳定性要求, 按照结构能量从低到高排序选取了15个团簇构型, 排除掉其中出现虚频的构型, 最终获得图3所示的13个团簇构型作为模拟谱计算的输入构型。
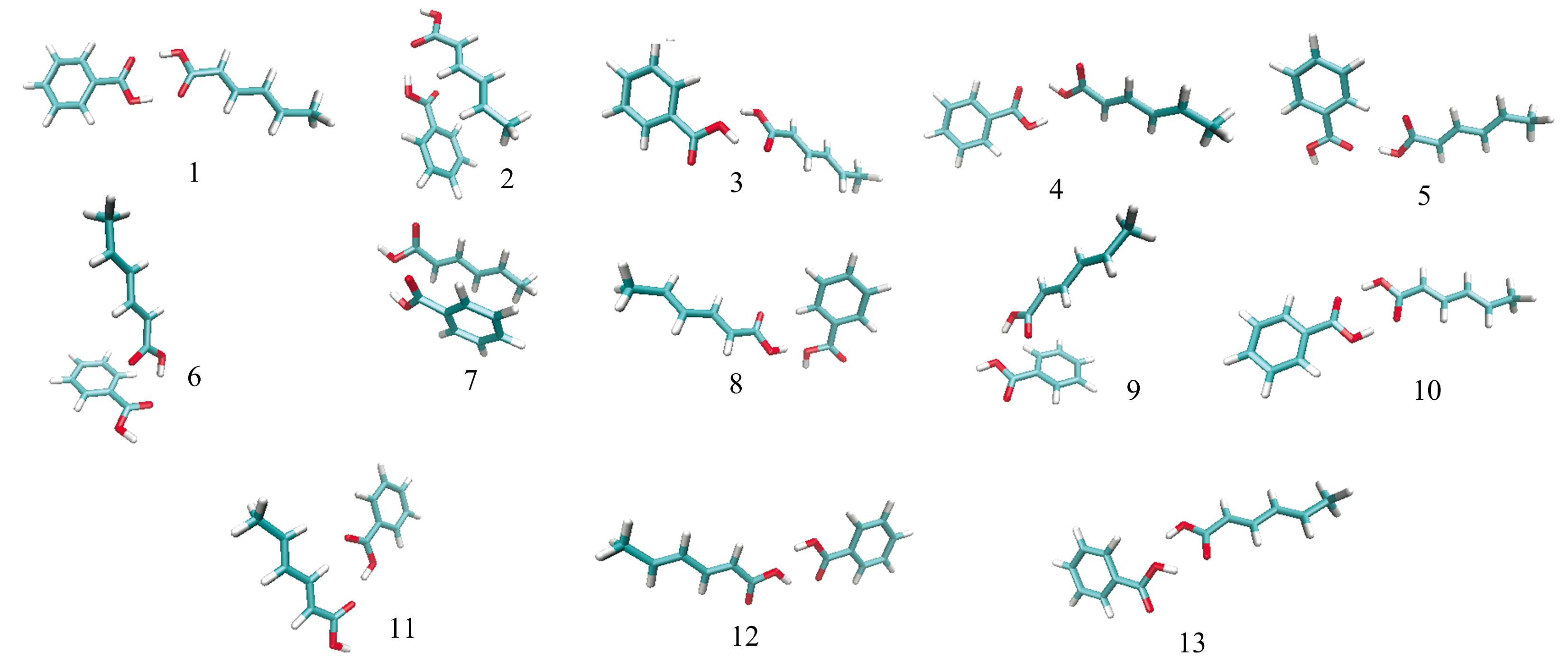 | 图3 苯甲酸和山梨酸混合物13个团簇构型Fig.3 Thirteen cluster configurations of benzoic acid and sorbic acid mixture |
利用Gaussian 09对上述的13个团簇构型分别在DFT-B3lyp-6-311g* * 水平下进行频率计算, 获取团簇构型在0.4~2.5 THz的模拟谱, 表2为13个团簇构型吸收峰的汇总信息。
| 表2 团簇构型Cluster1-13(sim)的吸收峰统计表 Table 2 Absorption peak statistics for cluster structures Cluster1-13(sim) |
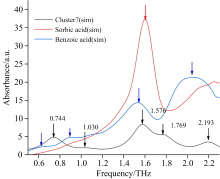
由图1可知, 苯甲酸和山梨酸混合物实验谱中包含4个吸收峰, 由表2可知, Cluster2(sim)、 Cluster5(sim)、 Cluster10(sim)构型的模拟谱吸收峰个数与之前图1二者混合物实验谱中吸收峰数量吻合, 但根据二者分别的模拟谱可知, 二者混合物的吸收谱应包含5个特征吸收峰, 表2中只有Cluster7(sim)构型的模拟谱具有5个吸收峰, 且峰位分别为0.744、 1.030、 1.576、 1.769和2.193 THz, 与实验谱峰位基本吻合, 如图4所示。 故因此推断Cluster7(sim)团簇构型更接近实际混合物样品的晶体结构。
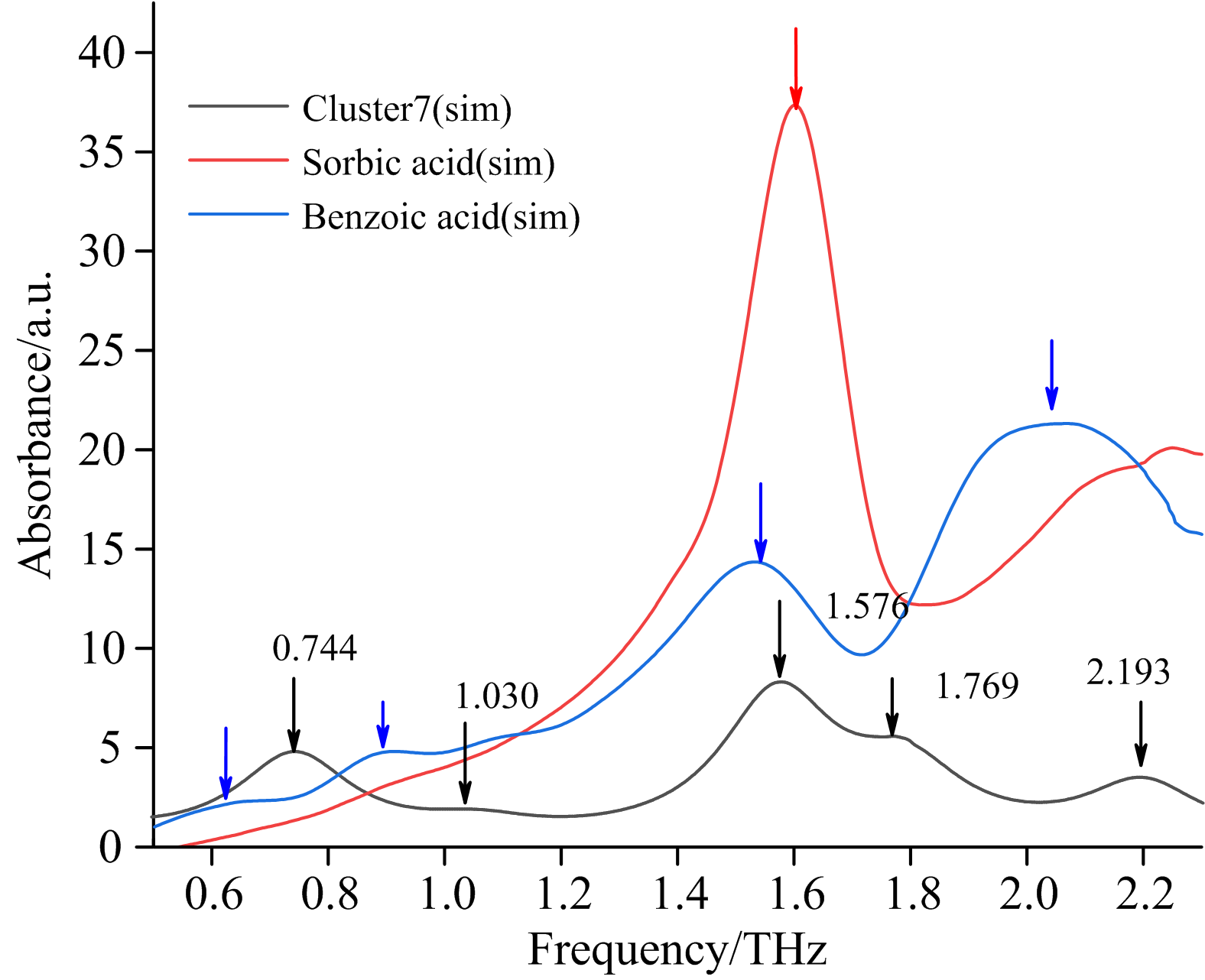 | 图4 基于混合物7号团簇构型的模拟谱与实验谱对比Fig.4 Comparison of simulated and experimental spectra based on the Cluster7(sim) configuration of the mixture |
有研究指出, 太赫兹范围内的特征吸收峰是由单个分子或晶体单元的低频振动引起的, 这些振动是由分子间或分子内部的键长、 键角和扭转角的变化所致。 且振动模式和旋转方式的不同也会影响吸收峰的位置和强度。 势能分布(PED)是一种分析方法, 用于分解分子的振动模式, 能够确定各基团在振动模式中的贡献值, 从而加深对振动模式特征的理解, 并帮助识别出主导的振动模式。 本文使用VEDA4对以上研究的晶胞构型的模拟谱吸收峰的振动模式进行PED分析, 完成模拟谱特征吸收峰的振动模式匹配[16]。 计算所得的苯甲酸、 山梨酸、 与Cluster7 (sim)团簇构型贡献率和振动模式见表3和表4。 表中, STRE表示键长伸缩, BEND表示键角弯曲, TORS表示二面角扭转, 括号内的数字为该振动模式占有的贡献百分比。 通过利用VMD[17]软件显示分子的振动分量, 可更直观地显示苯甲酸和山梨酸晶胞内各个原子的振动方向, 如图5所示。 图中黄色箭头表示苯甲酸和山梨酸分子分别在特征吸收峰处的振转模式。 由图5(a)可知, 苯甲酸在0.627 THz处的振动模式经分析可指认为贡献率为14%的46O45H所在平面的键长伸缩。
| 表3 苯甲酸、 山梨酸及其团簇构型振动模式PED分析 Table 3 PED analysis of benzoic acid and sorbate vibration modes |
| 表4 苯甲酸和山梨酸Cluster7(sim)团簇构型振动模式PED分析 Table 4 PED analysis of Benzoic acid and sorbate Cluster7(sim) cluster configuration vibration mode |
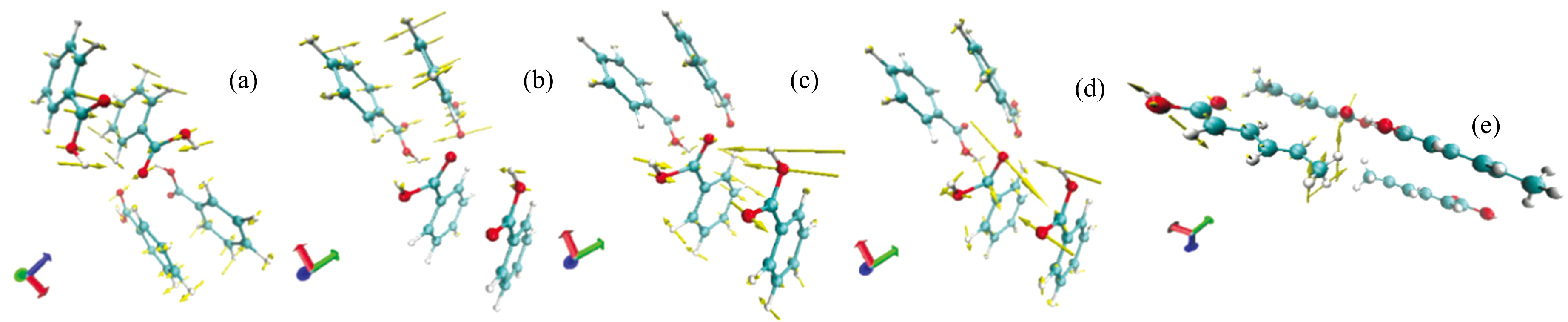 | 图5 振动模式 (a)、 (b)、 (c)、 (d)分别为苯甲酸在0.627、 0.980、 1.320和1.967 THz处的振动模式; (e)为山梨酸在1.446 THz处的振动模式Fig.5 Vibration modes (a), (b), (c) and (d) of benzoic acid at 0.627, 0.980, 1.320, 1.967 THz, respectively; Vibration modes (e) of sorbic acid at 1.446 THz |
由表3、 表4和图5发现苯甲酸和山梨酸的太赫兹吸收峰是复杂的分子内和分子间相互作用和各自分子的多种振动模式共同作用的结果。 这些模式包括羧基之间的氢键伸缩和弯曲振动, 羧基的C=O和C— O键伸缩振动, 苯环的呼吸和扭转振动, 以及C=C双键的伸缩振动、 C— H键的伸缩与弯曲振动, 还有O— H…O氢键间的键角伸缩和弯曲。 这些分子的振动模式可以通过吸收峰的位置信息进行区分。 通过对比两者的吸收峰位置和强度, 可以对吸收峰形成机理实现有效解析。
为了进一步考察分子内外相互作用, 解析特征吸收峰形成机理, 本文利用基于Hirshfeld原子空间划分的独立梯度模型(IGMH)对苯甲酸和山梨酸分子之间和内部的相互作用类型、 位置等进行了可视化分析。 相比于约化密度函数(reduced density gradient, RDG), IGMH更加直观且可以自由选取片段, 分析特定片段间或片段内的相互作用, 避免无关区域的等值面妨碍, 容易获得高效果的等值面, 耗时也更低[18, 19]。 IGMH函数表达式如式(3)和式(4)
式中, r为坐标矢量, i为原子序号, ∇为梯度算子, ρ i代表i原子的电子密度, |∇ρ i(r)|为取矢量的模。 δ g可以对结构中各原子间特定片段之间(interfragment)和内部(intrafragment)的相互作用进行直观地展现, 并将其分割为δ ginter和δ gintra。
通过将ρ (r)与电子密度Hessian矩阵中的第二大本征值λ 2的符号函数sign(λ 2)相结合, 所得的乘积sign(λ 2)ρ (r)可以映射到具有不同颜色的IGMH等值面上, 以此直观地揭示出共价作用、 弱相互作用的具体方位、 类型等[20]。 色彩梯度设定为蓝色过渡到绿色, 再过渡到红色, 其中蓝色代表ρ (r)> 0, λ 2< 0的区域, 这通常代表存在吸引作用; 绿色部分代表ρ (r)≈ 0, λ 2≈ 0的位置, 一般为弱氢键作用或范德华作用; 红色代表λ 2大于零的区域, 这通常关联于笼状结构或环状结构中较强的立体阻碍效应, 即位阻作用。 图6和图7展示的用波函数分析软件Multiwfn[21]和VMD计算得到的结果。
 | 图6 (a)苯甲酸填色IGMH散点图; (b)苯甲酸填色IGMH等值面图Fig.6 (a) Benzoic acid coloring IGMH scatter diagram; (b) benzoic acid coloring IGMH contour map |
 | 图7 (a)山梨酸填色IGMH散点图; (b)山梨酸填色IGMH等值面图Fig.7 (a) Sorbic acid coloring IGMH scatter diagram; (b) Sorbic acid coloring IGMH contour map |
图6(a)显示了苯甲酸晶胞的IGMH散点图, 图6(b)为其IGMH等值面图。 通过与PED分析结合, 针对苯甲酸吸收峰成因的特定原子进行了IGMH分析。 可以看出, 图中出现了多个不同位置的尖峰, 蓝色尖峰位于-0.60~-0.20 a.u, 体现出成键区域的电子密度较大, 这是共价键作用区域的典型特征。 结合图6, 可以推测该现象主要由C— C键(如19C— 24C、 20C— 21C、 34C— 39C、 35C— 36C等)、 C— H键(如20C— 25H、 34C— 40H等)及羧基中的C=O双键(48C=46O、33C=31O等)和其他共价键作用(如33C…32O— 45H、 18C…17O— 30H)导致。
在-0.04~0.04 a.u区域, 出现了较小的绿色尖峰, 这是弱相互作用区域, 主要归因于羧基中的O— H键和C=O双键与周围分子中的C— H键形成的弱氢键作用(如48C=46O…25H、18C=16O…55H、32O— 45H…44H等)。 此外, 苯环之间还存在π — π 堆积相互作用, 这是分子间芳香环之间的π 电子云相互吸引所导致的。
同时, 绿色区域也反映了范德华力的贡献, 尤其是在氢键和π — π 堆积较弱的区域, 范德华色散力通过分子间的瞬时偶极相互作用进一步增强了分子间的吸引力。 IGMH等值面图中对应化学键的区域周围有红色部分, 但这些区域没有明确的物理意义。
图7(a)为山梨酸晶胞IGMH散点图, (b)为山梨酸晶胞IGMH等值面图。 结合PED分析可知, 其山梨酸位于1.603 THz的太赫兹吸收峰成因可归结为: 羧基的C=O双键作为氢键受体与周围分子的C— H形成较弱的氢键(48C=35O…25H 等)作用, 这些氢键较弱, 但仍对分子间的空间排布和晶体的稳定性产生了一定影响。 其次, 山梨酸的分子骨架中存在多个C=C双键(如0C=42C), 这些作为共价键不仅维持了分子本身的稳定结构, 还通过分子振动参与了特定频率的太赫兹吸收。 与此同时, 山梨酸晶胞中的C— H键(42C— 43H、 44C— 45H、 46C— 47H)和羧基(48C…33O— 34H)也起到了共价键的作用, 进一步增强了分子结构的刚性。 分子间的范德华作用也广泛存在, 增强了晶体结构的稳定性。 IGMH图中的绿色区域代表了这些弱氢键和范德华力的作用, 它们共同维持了山梨酸晶体的有序性。
虽然IGMH对上述苯甲酸和山梨酸的晶胞构型进行了相互作用的可视化分析, 但在相同的等值面数值设置下难以清晰展示各种强度的相互作用, 而且需要高时长的计算。 故本文引入相互作用区域指示函数(IRI), 其既可以反映分子间的相互作用, 又可以反映出原子间的化学键[22]。 IRI 的函数定义如式(5)所示
式(5)中; ρ (r)是电子密度, ∇ρ (r)是梯度矢量, α 是可变参数。 与IGMH分析相似, IRI方法也是通过将sign(λ 2)ρ (r)函数在IRI等值面用不同颜色映射, 从而实现对不同区域间相互作用的识别。 因此, 色彩的参考标准与上述IGMH 是相同的。
图8是苯甲酸和山梨酸混合物Cluster7(sim)团簇构型的IRI填色散点图和等值面图。 从图8可以看出, 两个单分子羧基之间交互相错, 在此区域中C、 H、 O之间形成了不一的弱相互作用, 参见图8(b)中大面积的黄绿色等值面。 苯甲酸单分子羧基上的15H与山梨酸单分子羧基的16O间等值面也为绿色, 说明存在弱氢键作用, 31C=18O…28H、3C=1O…10H、2O— 15H…14H等之间呈现绿色和红色并存的区域, 说明同时存在吸引的弱氢键作用和一定程度的位阻效应。 IRI等值面中位于C— C键、 C— H键和C=O键之间的蓝色圆柱状物, 表示此处存在强的相互作用, 即化学键作用。 苯环中的红色区域说明存在较强的位阻效应, 且两个单分子间出现的大片微红绿色, 说明此处存在范德华作用。
 | 图8 (a)Cluster7(sim)团簇构型填色IRI散点图; (b)Cluster7(sim)团簇构型填色IRI等值面图Fig.8 (a) Cluster7(sim) configuration coloring IRI scatter diagram; (b) Cluster7(sim) configuration coloring IRI contour map |
结合团簇振动模式分析可知, 混合物团簇构型在1.576 THz处出现的吸收峰, 可归因于分子之间的氢键振动引起的, 其大部分振动出现在两个分子羧基之间的16O和15H的氢键上, 由于团簇构型改变了羧基的键长, 导致该频段出现了吸收峰。
首先利用透射式 THz-TDS系统结合光学参数提取得到了苯甲酸、 山梨酸及二者混合物在0.5~2.2 THz的吸收谱。 结果表明, 两种食品添加剂的吸收峰在峰位及峰数上均有显著区别, 故可基于太赫兹吸收谱完成两种添加剂的定性鉴别。 其次, 为了进一步解析特征吸收峰的形成机理, 明确混合物吸收谱中吸收峰缺失现象的原因, 使用Gaussian 09W软件, 将B3LYP结合DFT-D3进行校正, 对苯甲酸和山梨酸的晶胞结构进行了优化, 并通过Genmer构建是二者混合物的初始团簇构型, 后利用MOPAC、 Gaussian 09W对其进行结构优化和频率计算, 最后分别获取了两种添加剂的单质及混合物的太赫兹模拟谱, 结合相关软件对不同构型下的吸收峰振动模式进行了指认, 明确了特征吸收峰的主要振动方式。 最后, 结合Multiwfn和VMD软件, 使用IGMH和IRI分析对苯甲酸、 山梨酸晶胞及其混合物团簇构型中的相互作用进行了可视化分析, 解析了特征吸收峰的形成机理和振动模式的本质特征, 并分析了混合物太赫兹实验谱中吸收峰缺失的原因。 综上所述, 本文聚焦于分子光谱特性的基础研究, 探索了太赫兹频段内小分子及其混合物的特征吸收谱形成机制和分子间相互作用规律。 研究结果为太赫兹技术在分子光谱学领域的应用提供了理论与实验参考。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|