{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
过氧麦角甾醇振动光谱的理论计算与实验研究
[李一诺1  , 梁小蕊
, 梁小蕊1, * , 张纪磊1 , 李荫1 , 李晓栋2 ]
, 梁小蕊, 张纪磊|
|


作者简介: 李一诺, 2003年生,海军航空大学航空基础学院本科生 e-mail: 2867707940@qq.com; 梁小蕊, 1979年生,女,海军航空大学航空基础学院副教授 e-mail: xiaoruiliang12@163.com
过氧麦角甾醇是一种具有抗癌、 抗炎等多种生物活性的甾体衍生物, 在海洋生态系统中还具有一定的抗菌活性, 分析过氧麦角甾醇的结构对探究其活性机制至关重要。 密度泛函理论作为一种重要的量子化学计算方法, 已经越来越广泛地应用于预测分子的结构、 能量、 前线分子轨道以及有机结构光谱分析等领域。 利用GaussView 6.0软件构建过氧麦角甾醇分子的空间构型, 基于密度泛函理论DFT-B3LYP方法, 首先在Gaussian 09W软件中用3-21G基组进行初始结构粗优化, 在粗优化结构的基础上用6-311++G(d, p)基组进行结构的再优化, 得到分子的最稳定构型、 能量及前线轨道分布。 然后在优化结构的基础上, 选用6-311G基组计算了过氧麦角甾醇的理论红外光谱和拉曼光谱, 理论计算结果误差频率校正因子选择0.961 3进行修正, 并通过实验方法测得了过氧麦角甾醇固体粉末的红外光谱和拉曼光谱。 从理论计算和实验结果可以看出, 理论红外光谱中过氧麦角甾醇分子主要在3 700~2 800与1 500~600 cm-1范围两个区域有明显振动, 前者主要为伸缩振动, 后者包含多种振动类型。 理论与实验红外光谱的特征峰频率误差均小于30个波数, 说明理论计算结果较为可靠。 理论拉曼光谱中2 966~2 879 cm-1波段与实验光谱2 978~2 856 cm-1波段对应, 为C—H伸缩振动特征峰, 理论拉曼光谱的峰位较实验光谱略有蓝移, 整体峰位吻合较好。 该研究分析了过氧麦角甾醇的最优构型、 前线分子轨道及其振动光谱, 为过氧麦角甾醇的振动光谱检测与结构鉴定提供了理论基础, 为进一步探究其在海洋生态系统及医药领域的应用提供了基础的结构及光谱数据。
Ergosterol Peroxide is a steroid derivative with various biological properties such as anti-cancer, anti-inflammatory, etc. It also has certain antibacterial activity in marine ecosystems. Therefore, analyzing the structure of ergosterol peroxide is crucial for exploring its activity mechanism. As an important quantum chemical calculation method, density functional theory has been increasingly applied in predicting the structure, energy, frontier molecular orbitals, and organic structure spectroscopic analysis of molecules. In this work, the spatial structure of ergosterol peroxide molecular was constructed using GaussView 6.0 software. Based on the density functional theory DFT-B3LYP method, the initial structure of ergosterol peroxide was initially optimized using the 3-21G basis set in Gaussian 09W software. Based on the coarse optimized structure, the structure was further optimized using the 6-311++G (d, p) basis set to obtain the molecule's most stable configuration, energy, and frontier orbital distribution. Then, based on optimizing the structure, the theoretical infrared (IR) and Raman spectra of ergosterol peroxide were calculated using the 6-311G basis set. The error frequency correction factor of the theoretical calculation results was selected as 0.961 3 for correction. The experimental IR and Raman spectra of ergosterol peroxide solid powder were measured using experimental methods. From theoretical calculations and experimental results, it can be seen that in the theoretical infrared spectrum, ergosterol peroxide moleculesmainly exhibit significant vibrations in the range of 3 700~2 800 and 1 500~600 cm-1. The former mainly exhibits stretching vibrations, and the latter contains multiple vibrations. The characteristic peak frequency wavenumber error of both theoretical and experimental infrared spectra isless than 30 wavenumbers, indicating that the theoretical calculation results are relatively reliable. The corresponding bands in the theoretical Raman spectrum from 2 966 to 2 879 cm-1 and the experimental spectrum from 2 978 to 2 856 cm-1 are assigned to C—H stretching vibration characteristic peaks. The peak positions in the theoretical Raman spectrum are slightly blue-shifted compared to the experimental spectrum, but overall, they agree well. This study analyzed the optimal structure, frontier molecular orbitals, and vibration spectra of ergosterol peroxide, providing a theoretical basis for vibration spectrum detection and structure identification. Fundamental structural and spectral data were provided to further explore the application of ergosterol peroxide in marine ecosystems and the pharmaceutical field.
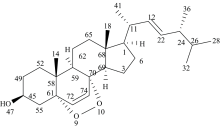
过氧麦角甾醇(ergosterol peroxide, EP)是一种甾体衍生物, 为无色晶体, 常见于多种真菌、 酵母、 地衣或海绵中。 其化学名称为5α , 8α -过氧化麦角甾-6, 22-二烯-3β -醇, 化学式为C28H44O3, 分子结构如图1[1, 2]。 过氧麦角甾醇具有显著的抗癌活性以及抗炎、 抗病毒和免疫调节等多种生物活性, 在抗癌药物的研究方面具有重要价值。 同时, 过氧麦角甾醇在海洋生态系统中具有一定的抗菌活性, 可以作为海洋细菌的抑制剂[3, 4, 5, 6, 7]。 因此, 对于过氧麦角甾醇的研究在海洋生态系统及医药领域具有重要意义。 目前, 对于过氧麦角甾醇的研究主要集中在分离纯化、 含量测定, 以及抗癌活性表征等方面, 对其几何构型、 前线轨道、 振动光谱等的研究鲜有报道, 这些结构性质是决定其抗癌、 抗菌活性的重要因素。
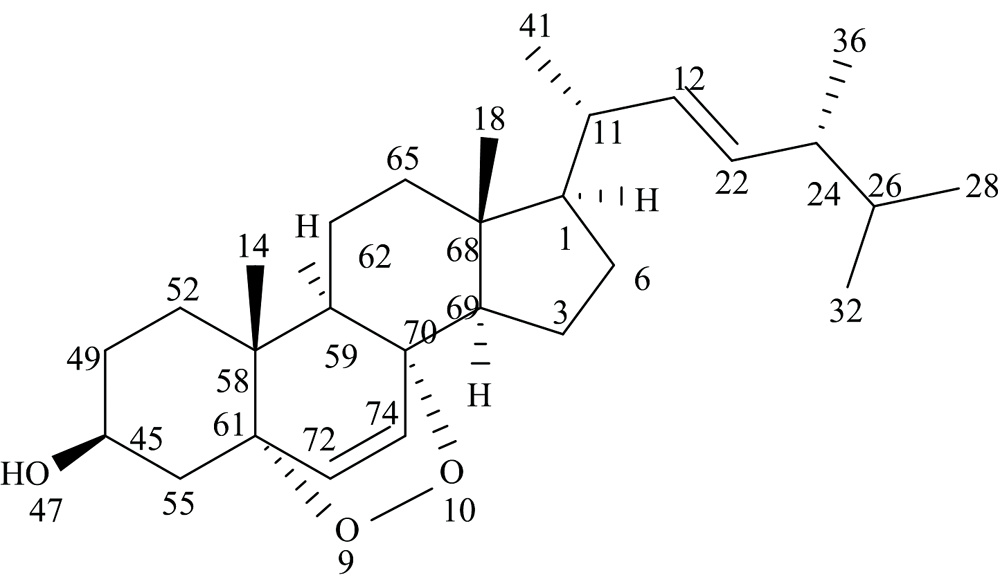 | 图1 过氧麦角甾醇分子平面结构Fig.1 Planar structure of ergosterol peroxide |
随着现代分析仪器和计算机技术的发展, 量子化学计算在化学结构分析领域起到了越来越重要的作用。 密度泛函理论(density functional theory, DFT)是一种研究多电子体系结构的量子化学计算方法, 能预测分子的稳定几何结构、 能量、 振动光谱等性质[8, 9, 10]。 本工作利用Gaussian 09W软件包, 基于密度泛函理论(DFT), 对一个从真菌棘孢木霉的次生代谢产物中分离得到的过氧麦角甾醇化合物分子进行了结构全优化, 在优化稳定构型的基础上绘制了过氧麦角甾醇分子的理论红外光谱和拉曼光谱, 同时实验测定了其红外光谱和拉曼光谱。 分析探讨了过氧麦角甾醇分子的几何构型、 前线轨道, 并将两种光谱的理论计算结果与实验结果进行了比较分析, 为进一步研究过氧麦角甾醇的活性机制、 结构鉴定提供了有用的信息和理论依据。
过氧麦角甾醇是从一株真菌棘孢木霉的次生代谢产物中分离纯化得到的, 该棘孢木霉来源于海洋褐藻马尾藻[11]。 红外光谱(IR)的测定采用日本JASCO FT/IR-4100红外光谱仪; 拉曼光谱测试采用DXR RamanMicroscope型激光拉曼光谱仪, 选择波长532 nm激光作为激发光源。
过氧麦角甾醇的分子构型由GaussView 6.0软件构建, 量子化学理论计算采用Gaussian 09W单机版软件包。 计算基于DFT方法的B3LYP泛函, 首先选用3-21G基组, 对EP分子进行几何结构粗优化, 在得到粗优化构型的基础上, 选择6-311++G(d, p)基组进行再优化, 并在优化后稳定构型的基础上, 选用6-311G基组计算了分子的理论红外光谱与拉曼光谱。 通常量化计算把所有振动都看作简谐振动而使计算频率较实验值偏大, 因此, 理论光谱的频率修正选择文献报道的0.961 3作为校正因子[12]。
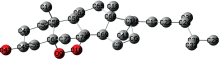
利用Gaussian 09W软件, 基于DFT-B3LYP方法, 对EP分子进行几何结构全优化, 图2为优化后的几何构型。 EP分子为甾族化合物, 以四环碳骨架甾核为主体, 45C上连有一个羟基, 61C和70C上连有一个过氧基, 72C和74C形成双键。 表1、 表2、 表3分别为EP分子的键长, 键角和二面角参数。
 | 图2 B3LYP/6-311++G(d, p)优化后的过氧麦角甾醇分子立体构型Fig.2 Optimized configuration of ergosterol peroxide at B3LYP/6-311++G(d, p) levels |
| 表1 优化后过氧麦角甾醇分子的键长参数 Table 1 Bond lengths of ergosterol peroxide obtained by configuration optimization |
| 表2 优化后过氧麦角甾醇分子的键角参数 Table 2 Bond angles of ergosterol peroxide obtained by configuration optimization |
| 表3 优化后过氧麦角甾醇分子的二面角参数 Table 3 Dihedral angles of ergosterol peroxide obtained by configuration optimization |
由表1键长数据可知, 61C— 58C键长为1.569 01 Å , 59C— 70C键长为1.556 68 Å , 59C— 58C键长为1.575 69 Å , 均大于未取代六元环的键长, 70C— 74C键长为1.506 97 Å , 74C— 72C键长为1.334 38 Å , 72C— 61C键长为1.509 36 Å , 均小于未取代六元环的键长, 说明由于过氧基所在六元环的存在, 使58C— 61C— 72C所在的六元环产生了较大的结构变化。 49C所在的六元环, 其键长均大于未取代的六元环, 说明受61C所在的3个六元环的影响, 49C的六元环略有放大。 而61C— 58C单键是49C所在的六元环与59C、 58C、 61C所在的两个六元环共用的碳碳单键, 因此, 受49C所在六元环的影响, 其键长有所增加。
由表2键角数据可知, ∠24C— 22C— 12C为126.497 14° , ∠22C— 12C— 11C为129.104 36° , 均与sp2杂化轨道120° 有较大差距, 说明22C上的取代基影响了双键的空间结构。 ∠70C— 74C— 72C为113.265 05° , ∠74C— 72C— 61C为113.654 02° , 说明由于过氧基的取代形成了三个六元环, 对74C=72C双键产生了较大影响。
由表3二面角数据可知, ∠74C— 72C— 61C— 9C为55.984 29° , ∠10C— 9C— 61C— 72C为-62.188 07° , ∠58C— 59C— 70C— 10C为52.535 6° , 说明过氧基取代形成的三个六元环均不在同一平面内, 空间结构较为复杂。
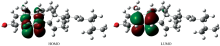
前线轨道理论(frontier molecular orbitals, FMO)由Fukui[13]在20世纪50年代提出, 是一种分子轨道理论, 分子轨道中的最高占据分子轨道(HOMO)和最低空分子轨道(LUMO)及二者的能级差可以用来判断分子的稳定性, 并为确定分子化学活性部位及探讨反应机理等提供有用的信息[14, 15, 16]。 利用Gaussian 09W软件, DFT-B3LYP方法计算得到的EP分子优化构型的能量为: -35 901.59 eV(-1 319.358 345 a.u), 能量较低, 说明分子为稳定状态。 HOMO能量为-8.298 352 eV(-0.304 959 a.u.), LUMO能量为-4.919 392 eV(-0.180 784 a.u.), 能隙为3.378 960 eV(0.124 175 a.u.)。 能隙的大小能够反映电子发生轨道跃迁的能力, 能隙越小, 电子越容易发生轨道跃迁, 一般能隙不超过6ev的分子容易发生电子跃迁, 计算结果说明EP分子具有一定的电子跃迁能力。 利用GaussView可视化软件绘制了EP分子的HOMO和LUMO电子云分布图, HOMO和LUMO轨道类型均属于alpha+beta型轨道, 如图3所示。 由图可知HOMO轨道电子云主要分布在59C、 58C、 61C、72C=74C双键, 以及9O— 10O过氧原子上, LUMO轨道电子云主要分布在70C— 59C— 58C— 61C— 72C=74C六元环和9O— 10O过氧原子上。 将优化后的EP分子稳定结构在Multiwin软件中进行轨道组成分析, 分别计算出每个原子对分子前线轨道的贡献率[17, 18, 19], 如表4所示。 由表4可见, EP分子中的9O、 10O对HOMO轨道的贡献率最大, 分别为41.352 69%和41.703 91%; 对LUMO轨道贡献率最大的原子为72C和74C, 分别为40.254 85%和40.120 94%, 这与GaussView计算得到的HOMO和LUMO电子云分布图是一致的。 HOMO和LUMO还可用来预测分子的亲核反应和亲电反应, HOMO与亲电反应有关, LUMO与亲核反应有关[15]。 由表4数据可知, EP分子中的9O、 10O对HOMO的贡献率远远大于其他原子的贡献率, 因此, 这两个氧原子最易受到亲电试剂的攻击, 发生亲电反应; 而在LUMO中, 72C和74C的贡献率远远大于其他原子, 因此72C和74C最易受到亲核试剂的攻击发生亲核反应。
 | 图3 过氧麦角甾醇分子的HOMO和LUMO图Fig.3 The highest occupied molecular orbital and the lowest unoccupied molecular orbital of ergosterol peroxide molecule |
| 表4 各原子对过氧麦角甾醇分子前线轨道的贡献 Table 4 Contribution of atoms to the frontier molecular orbitals of ergosterol peroxide |
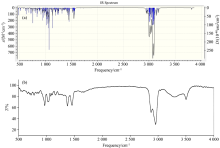
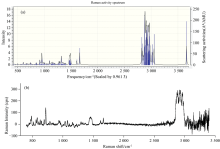
红外光谱(infrared spectroscopy, IR)是利用物质分子对红外辐射的吸收, 由其振动或转动引起偶极矩的净变化, 使分子振动能级和转动能级从基态跃迁到激发态, 得到分子振动能级和转动能级变化产生的振动-转动光谱。 IR具有用量少、 分析速度快、 不破坏样品等优点, 在鉴定未知物结构或确定官能团方面应用广泛[20]。 在优化结构基础上利用DFT-B3LYP方法, 6-311G基组, 对EP分子的频率进行了计算, 结果收敛且无虚频。 通过GaussView 6.0软件观察EP分子的理论红外光谱的振动形式, 绘制了气相条件下EP分子在中红外区4 000~500 cm-1波段范围内的红外光谱图, 如图4(a)所示。 图4(b)为实验测得的EP固体粉末纯品的红外光谱。
 | 图4 过氧麦角甾醇分子的理论红外谱图(a)和实验红外谱图(b)Fig.4 Theoretical IR spectrum (a) and experimental IR spectrum (b) of ergosterol peroxide |
由图4理论和实验红外光谱可见, EP分子的红外振动吸收峰主要集中在两个波段区: 3 700~2 800 cm-1和1 500~600 cm-1。 其中3 700~2 800 cm-1区域主要归属于伸缩振动, 1 500~600 cm-1波段区的振动较为复杂。 实验红外光谱中3 500 cm-1附近一个较宽的吸收峰是溴化钾中水的O— H的吸收峰[21]。 整体来看红外理论光谱与实验光谱在特征吸收峰上吻合较好, 说明用DFT-B3LYP方法可以有效地研究EP分子, 并对甾醇类化合物的光谱预测具有一定的指导作用。
结合理论计算和实验数据对图4振动模式进行分析可知, 3 700~2 800 cm-1波段内, 理论红外光谱在3 664.26 cm-1处的极弱吸收峰为EP分子中羟基的O— H伸缩振动, 这一吸收峰在实验光谱中被包含在溴化钾水峰中而未有体现; 计算数据中2 966.30、 2 962.43、 2 947.70、 2 946.66、 2 940.68、 2 889.58、 2 879.05 cm-1处的中强或强吸收峰为EP分子中甲基的C— H伸缩振动峰, 2 958.90、 2 956.55、 2 954.74、 2 907.08 cm-1处的中强吸收峰为EP分子中各六元环和五元环中C— H的伸缩振动峰, 与实验振动频率2 983~2 859 cm-1基本相符。 1 500~600 cm-1波段内, 理论光谱中1 490.05 cm-1处的弱吸收峰主要是28C、 32C、 36C所在甲基的C— H不对称变形振动, 与实验值中的1 459.85 cm-1基本一致; 1 463.39和1 195~1 190 cm-1处的弱吸收峰主要是各环的亚甲基C— H变形振动及骨架振动, 与实验值中的1 448和1 160~1 155 cm-1基本一致; 1 397.82 cm-1处的中强吸收峰和1 393.79 cm-1处的弱吸收峰主要是几个甲基的C— H对称变形振动, 对应实验值为1 378.85、 1 375.00 cm-1; 1 031.09 cm-1处的中强吸收峰是羟基O— H的变形振动; 994.58 m-1处的弱吸收峰主要是12C=22C (反式)的C— H变形振动, 对应实验值为968.09 cm-1; 1 097.04 cm-1处的中强吸收峰和731.63 cm-1处的弱吸收峰为72C=74C(顺式)的C— H变形振动, 对应实验值为1 076和715 cm-1。 上述红外光谱的理论值与实验值的误差均小于30个波数, 二者吻合很好, 说明用DFT-B3LYP方法, 在6-311G基组水平上计算EP分子的红外光谱是可靠的。
拉曼光谱与红外光谱都是分子振动光谱, 但二者在工作原理、 测定方式和谱图解析等方面存在一定差异。 拉曼光谱不是观察光的吸收, 而是观察光的非弹性散射。 拉曼光谱能够提供丰富的分子振动信息, 因此被广泛应用于化学成分和结构的分析、 生物分子的定量检测、 环境样品的快速检测等[22, 23, 24]。 红外光谱和拉曼光谱均能够反映分子振动能级和转动能级的变化[20], 两种光谱结合来看可以得到较为准确的分子结构信息。 图5给出了EP分子的拉曼光谱。 其中图5(a)为用DFT-B3LYP/6-311G方法计算出的EP分子理论拉曼光谱, 图5(b)为实验测得的EP分子拉曼光谱。
 | 图5 过氧麦角甾醇分子的理论拉曼谱图(a)和实验拉曼谱图(b)Fig.5 Theoretical Raman spectrum (a) and experimental Raman spectrum (b) of ergosterol peroxide |
比较图5(a)和(b)可见, 理论拉曼光谱特征峰较实验光谱略有蓝移, 但整体峰位吻合较好。 如理论拉曼光谱中1 662.75和1 608.62 cm-1处的特征峰为碳碳双键的伸缩振动峰, 对应实验值为1 665.01和1 620.65 cm-1; 理论拉曼光谱中2 966~2 879 cm-1波段区域的C— H伸缩振动特征峰, 对应实验光谱在2 978~2 856 cm-1波段。
拉曼光谱与红外光谱在某些方面具有互补性, 拉曼光谱可以提供样品中特定化学键的振动模式和分子结构的信息, 而红外光谱则可以提供样品振动频率和分子结构更详细的信息。 两种光谱的形成原理不同, 同一种官能团在两种光谱中的体现并不完全相同。 例如在理论拉曼光谱中1 319.44、 1 484.04、 1 608.62和1 662.75 cm-1处的特征峰, 在红外光谱中观察不到; 2 952.34、 2 911.89、 2 873.21、 1 393.79、 1 031.09 cm-1处, 在红外光谱中有明显吸收峰, 但在拉曼光谱中观察不到。
选取从海洋来源真菌棘孢木霉的次生代谢产物中分离纯化得到的过氧麦角甾醇化合物作为研究对象, 基于DFT-B3LYP方法, 利用Gaussian 09W和GaussView 6.0程序, 对过氧麦角甾醇分子的初始结构进行了全优化, 绘制了过氧麦角甾醇分子的HOMO和LUMO电子云分布图, 分析了分子的最稳定结构和前线轨道。 通过分析分子的前线轨道可知, 过氧麦角甾醇分子能隙较小, 具有一定的电子跃迁能力; 通过分析各原子对HOMO、 LUMO轨道的贡献率得出过氧麦角甾醇分子中的过氧原子9O、 10O最易受到亲电试剂的攻击, 发生亲电反应; 六元环中的双键72C和74C最易受到亲核试剂的攻击发生亲核反应。
在优化构型的基础上, 选取6-311G基组, 计算了过氧麦角甾醇分子的理论红外和拉曼光谱, 将两种光谱进行了比较分析, 并通过GaussView 6.0软件对不同频率峰的振动模式进行了指认归属。 同时实验测定了过氧麦角甾醇固体样品的红外光谱和拉曼光谱, 并将实验结果与理论计算结果进行了对比分析, 发现理论与实验红外光谱和拉曼光谱的对应峰值吻合均较好, 说明基于DFT方法计算的量子化学模型准确可靠。 本研究可为过氧麦角甾醇的振动光谱检测和分子结构鉴定提供基础数据的储备。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|