


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
新型二萜内酯化合物Euphorikanin A的结构和性质计算分析
[徐佳禛1  , 王长江
, 王长江1 , 王朝杰2, * ]
, 王长江]
|
|


作者简介: 徐佳禛,女, 1993年生,嘉兴学院附属第二医院主管药师 e-mail: xujiazhen0529@163.com
化合物Euphorikanin A是从甘遂大戟根中分离出的新型二萜内酯化合物, 初步生物活性发现其具有肿瘤细胞毒活性, 分析药物的结构、 光谱性质和成药可能性是了解其生物活性的必要前提, 也可为开发新药提供参考。 运用密度泛函理论B3LYP/6-311++G(2d, p)和ωB97XD/6-311++G(2d, p)方法, 对化合物Euphorikanin A的药效构象、 几何和电子结构、 红外(IR)、 紫外-可见(UV-Vis)、 核磁共振(NMR)谱进行计算, 并借助概念密度泛函理论进行分子全局反应指数分析, 使用药物代谢动力学平台开展成药性和ADME/Tox评估。 计算结果显示Euphorikanin A有唯一药效构像, 两种方法和不同溶剂环境中化合物几何结构参数值相近, 计算值与晶体参数吻合较好。 理论红外光谱特征与实验吻合, 水环境中计算的IR光谱更接近真实环境, 理论计算校正因子为0.94。 Euphorikanin A紫外光谱最大吸收峰在193.88~201.75 nm。 两种方法得到的核磁数据理论结果与实验吻合度高, R2均大于0.94。 Euphorikanin A的六元环的羟基可能为反应活性位点。 Euphorikanin A的全局反应参数和药代动力学表现出良好性能, 尤其在Caco-2细胞膜通透性、 血脑屏障透过率和人肠道吸收性。 Euphorikanin A的结构和性质在成药性拥有优势, 具有进一步开发的价值。
Euphorikanin A, a recently discovered diterpene lactone compound is dated from the roots of Euphorbia reausui, exhibits promising tumor cytotoxic activity. To comprehend its biological effects fully, it is imperative to investigate its structural characteristics, spectral properties, and potential as a therapeutic agent. Consequently, further examination of its structure and properties can serve as a valuable resource for advancing novel pharmaceuticals. The density functional theory B3LYP/6-311++G (2d, p) and ωB97XD/6-311++G (2d, p) methods were applied to calculate the pharmacophoric conformation, geometric and electronic structure, infrared (IR), ultraviolet-visible (UV-Vis), nuclear magnetic resonance (NMR) spectra of the compound Euphorikanin A, and the molecular global reaction index analysis was carried out by using conceptual density functional theory. The pharmacokinetics platform was used to evaluate druggability and ADME/Tox. The calculation results showed that Euphorikanin A has a unique pharmacophoric conformation, and the geometric structure parameters of the compound were similar in both methods and different solvent environments. The calculated values were in good agreement with the crystal parameters. The theoretical infrared spectral characteristics are consistent with the experiment, and the calculated infrared spectra in the water environment are closer to the real values, with a theoretical scaling factor of 0.94. The maximum absorption peak of Euphorikanin A in the UV-Vis spectrum is between 193.88 and 201.75 nm. The theoretical results of the nuclear magnetic data obtained by both methods agree well with the experimental results, with R2 greater than 0.94. The hydroxyl group of the six-membered ring of Euphorikanin A may be the reaction's active site. The compound's global reaction parameters and pharmacokinetics showed good performance, especially regarding Caco-2 cell membrane permeability, blood-brain barrier permeability, and human intestinal absorption. The structure and properties of Euphorikanin A have advantages in druggability and are worthy of further development.
大戟科植物约有322属8 910种, 广泛分布于热带和亚热带地区。 大量研究报道了从大戟属植物中分离得到的二萜类化合物具有抗肿瘤、 细胞毒性、 多药耐逆转、 抗病毒特性、 多种血管效应、 抗炎活性等多种生物活性[1, 2, 3]。 2012年, 从大戟植物中提取的巨大戟醇甲基丁烯酸酯凝胶制剂被FDA批准用于治疗光化角质病[4]。 大戟科二萜类化合物普罗斯左汀可以激活潜伏的HIV病毒, 与现有抗HIV药物组合使用可能获得根除HIV感染的疗法, 普罗斯左汀还表现出显著抗肿瘤活性[5]。 树脂毒素(resiniferotoxin)是树脂大戟分泌的一种辣椒素类似物, 属瑞香烷型二萜, 有镇痛作用和其他重要活性, 正在进行临床二期、 三期的评估[6]。
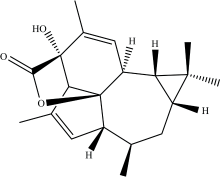
甘遂大戟, 俗称“ 甘遂” , 属于大戟科, 主要以甘遂的块根入药[7]。 2016年, 兰州大学张占欣团队从甘遂大戟的根中分离出了一种全新的二萜内酯化合物, 命名为Euphorikanin A, 结构如图1所示。 初步生物活性研究表明, 化合物Euphorikanin A对两种人类肿瘤细胞系NCI-446和HeLa具有中等细胞毒活性, IC50值分别为20.89和28.85 μ mol· L-1 [8]。 其结构Euphorikanin A具有高张力的5/6/7/3-稠合四环骨架, 带有桥接的[3.2.1]-γ -内酯, 并具有八个连续的手性中心。 Euphorikanin A新颖而复杂的分子骨架以及其潜在的生物活性使其成为全合成的优选目标。 2020年, 北京大学深圳研究生院的杨震课题组采用了连续多米诺式烯烃复分解反应一步构建了其中的五元环和六元环(可实现克级制备), 通过修饰完成Euphorikanin A主体骨架的合成[9]。 2021年, 瑞士联邦理工学院的Carreira课题组首次实现了Euphorikanin A的全合成, 其中的关键步骤为通过SmI2介导的内酯合成的同时实现五元环的关环[10]。 2022年, 北京大学药学院Chen等发表生物源合成途径启发的天然产物Euphorikanin A的全合成[11]。
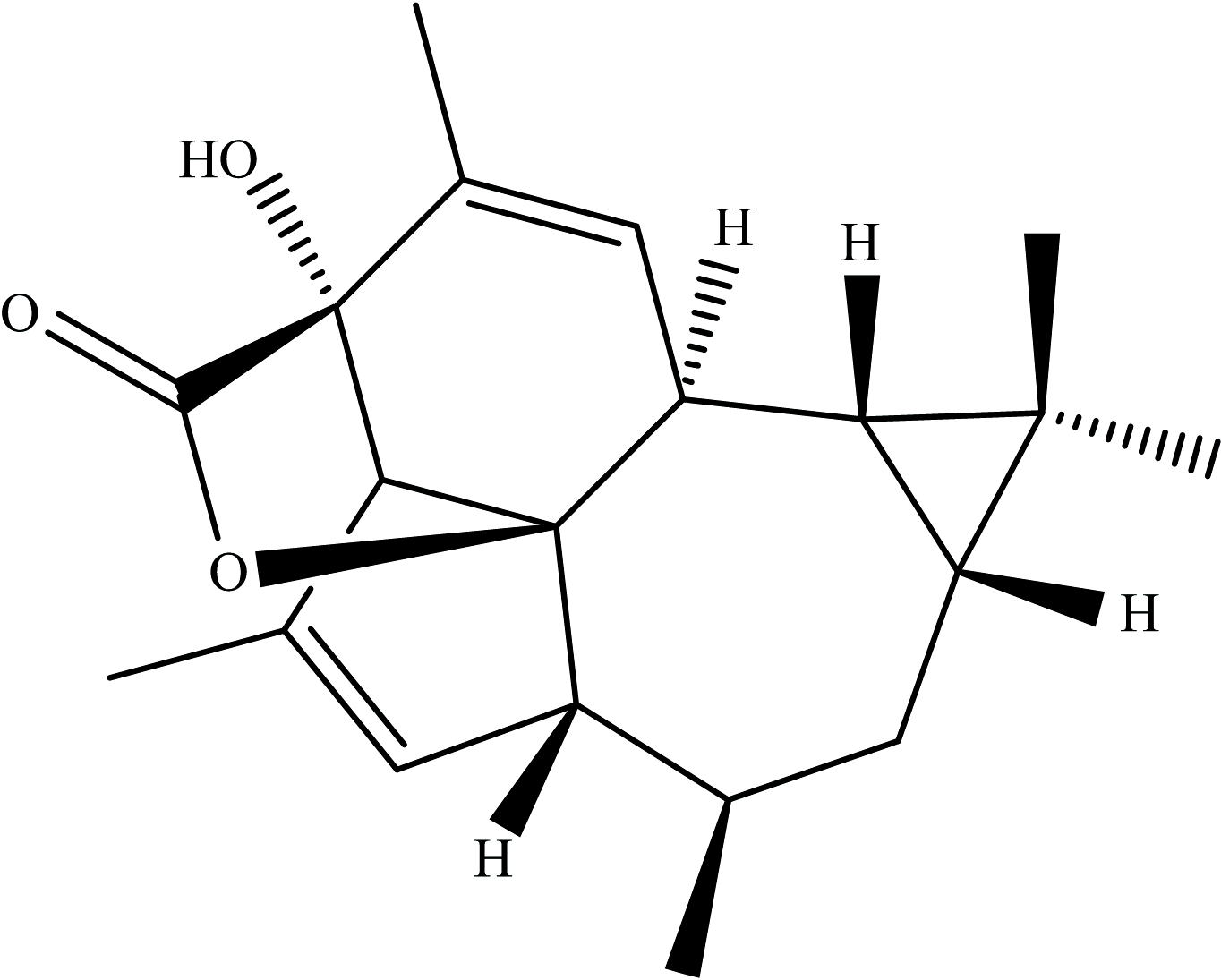 | 图1 化合物Euphorikanin A的2D结构式Fig.1 2D-structural formulas of Euphorikanin A |
化合物Euphorikanin A的研究主要集中在分离、 鉴定和全合成, 对其电子结构、 反应性和成药性的研究鲜有报道, 未见该化合物的计算研究。 了解药物的结构、 光谱性质和成药可能性是了解其生物活性的必要前提。 因此对化合物Euphorikanin A, 结构式见图1, 进行系统计算分析具有重要的意义。
分子构象采用MMFF94力场进行分子力学计算, 在GaussView6.0中的附加模块GMMX中实现。 使用默认参数集, 总能隙为3.5 kcal· mol-1。 当超过10 000次试验的限制, 或经过50次连续试验, 仍没有新的构象位于能量间隙内全局最低限度, 则停止搜索。
运用密度泛函理论的B3LYP和ω B97XD方法, 在6-311++G(2d, p)基组水平下对真空环(Vacuum, Vac)境中的Euphorikanin A结构进行优化, 得到稳定结构。 在稳定构型的基础上采用相同的理论方法对其电子结构、 红外(IR)振动光谱、 紫外-可见(UV-Vis)吸收光谱、 核磁共振(NMR)谱进行计算。 已有研究表明B3LYP和ω B97XD方法在研究化合物结构和光学性质时, 其计算结果与实验值吻合较好[12, 13, 14]。 在上述优化结构的基础上, 结合自洽反应场方法中的极化连续介质模型模拟水(water, Wat)、 环己烷(cyclohexane, Cyc)和甲醇(methanol, Met)等溶剂环境, 在同一基组水平进行上述性质的计算。
药物分子与受体之间的相互作用同大多数有机化学反应具有类似性, 可以通过分子的亲电性/亲核性和自由基性预测和解释其反应活性和反应位点[15]。 为了阐明分子中的化学键、 反应倾向和反应位点, 提出了电离势(IP)、 电子亲和势(EA)、 电负性(χ )、 整体硬度(η )、 化学势(μ )、 整体亲电指数(ω )和整体柔软度(S)等全局反应性描述符, 计算参照文献中定义公式[16]。 药代动力学计算借助ACD/Labs Percepta平台实现, 该平台基于QSAR(quantitative structure-activity relationship)模型预测化合物的药代动力学部分参数和理化性质。
所有量化计算和分析使用Guassian 16程序[17]、 Multiwfn 3.8软件[18]和VMD 1.9.3软件[19]完成。
在化合物Euphorikanin A中只有与羟基相连的C— O键可以旋转产生构像, 以及环构像变化。 运用MMFF94力场搜索显示Euphorikanin A分子只有1种构像。 由于具有高张力的5/6/7/3-稠合四环骨架, 带有桥接的[3.2.1]-γ -内酯部分, 并具有八个连续的手性中心且五元六元环均有双键, 限定了构象的变化数, 因而化合物Euphorikanin A的构像限制大大提高了其成药可能性[20]。
图2和表1为采用B3LYP/6-311++G(2d, p)和ω B97XD/6-311++G(2d, p)方法, 在真空、 水、 环己烷和甲醇环境中计算的化合物Euphorikanin A稳定分子构型和部分结构参数。
 | 图2 化合物Euphorikanin A优化后的稳定分子构型Fig.2 Optimized geometric structure of Euphorikanin A |
| 表1 化合物Euphorikanin A理论计算和实验的部分结构参数 Table 1 Experimental and theoretical comparison of Euphorikanin A geometry |
在真空中B3LYP/6-311++G(2d, p)计算水平化合物Euphorikanin A结构中C6=O8、 C6— O7、 C15— O16和C5— C15的键长分别为1.19、 1.37、 1.40和1.52 Å , 两种方法计算结果一致。 考虑极化连续介质模型后, C6=O8、C15— O16和C5— C15键长随着溶剂极性增大而缩短, 减少约0.01 Å 。 Fei等[8]从甘遂中分离Euphorikanin A化合物并通过X射线晶体结构测定得到的键长参数与本研究理论计算相差约0.02 Å 。 化合物Euphorikanin A中存在6个分子内氢键, 氢键键长在2.49~3.01 Å 范围内, 其中内酯环上的O7与五元环上的C5— H形成的分子内氢键相对较强。 与文献报道的其他分子内氢键强度相比, 化合物Euphorikanin A的分子内氢键较弱[21]。
化合物Euphorikanin A为5/6/7/3四环并环骨架不在同一平面内的刚性结构。 以真空环境中B3LYP方法计算为例, 五元环和六元环之间的键角(∠C3— C4— C10)为112.6° , 五元环和七元环之间的键角(∠C1— C3— C21)为117.3° , 六元环和七元环之间的键角(∠C12— C10— C13)为111.9° , 七元环和三元环之间的键角(∠C10— C12— C18)为121.7° , 两种方法和不同环境中计算得到的结果相差不大。
图3(a)与图3(b)分别为B3LYP/6-311++G(2d, p)和ω B97XD/6-311++G(2d, p)水平计算了真空、 水环境、 环己烷和甲醇环境下化合物Euphorikanin A的IR振动吸收光谱, 如图3(a, b)所示。 化合物Euphorikanin A在1 700~1 900 cm-1有强吸收峰, 由羰基的伸缩振动引起, 在3 750~3 900 cm-1范围内有羟基的特征峰。 计算的四种环境中的羰基伸缩振动频率分别为1 887、 1 792、 1 830和1 794 cm-1, 与文献报道的化合物Euphorikanin A羰基伸缩振动实验值(1 756 cm-1)相比呈蓝移现象。 羟基的伸缩振动分别为3 884、 3 813、 3 823和3 813 cm-1, 与实验值(3 442 cm-1)相比蓝移约391 cm-1 [8]。 在水环境中计算的IR光谱更接近真实环境, 理论计算的约化校正因子为0.94。
 | 图3 B3LYP/6-311++G(2d, p) (a)和ω B97XD/6-311++G(2d, p) (b)水平计算四种环境Euphorikanin A的IR吸收光谱Fig.3 The IR spectra of Euphorikanin A at the B3LYP/6-311++G(2d, p) (a) and ω B97XD/6-311++G(2d, p) (b) levels of theory in four environments |
化合物Euphorikanin A在真空、 水、 环己烷和甲醇环境中计算得到的UV-Vis吸收光谱曲线见图4(a, b)。 见图4(a)真空环境B3LYP/6-311++G(2d, p)基组水平计算得到的化合物Euphorikanin A紫外光谱最大吸收峰位于201.75 nm处, 该吸收峰主要包含52%的HOMO轨道电子向LUMO+7跃迁和10%的HOMO轨道电子向LUMO+5跃迁。 见图4(b)中ω B97XD方法计算的最大吸收峰位于193.88 nm, 与B3LYP方法相比蓝移7.87 nm。 考虑溶剂效应, B3LYP方法计算的化合物Euphorikanin A紫外光谱最大吸收峰红移约6.19 nm, ω B97XD方法计算的最大吸收峰蓝移约8.67 nm。 实验数据未见报道。
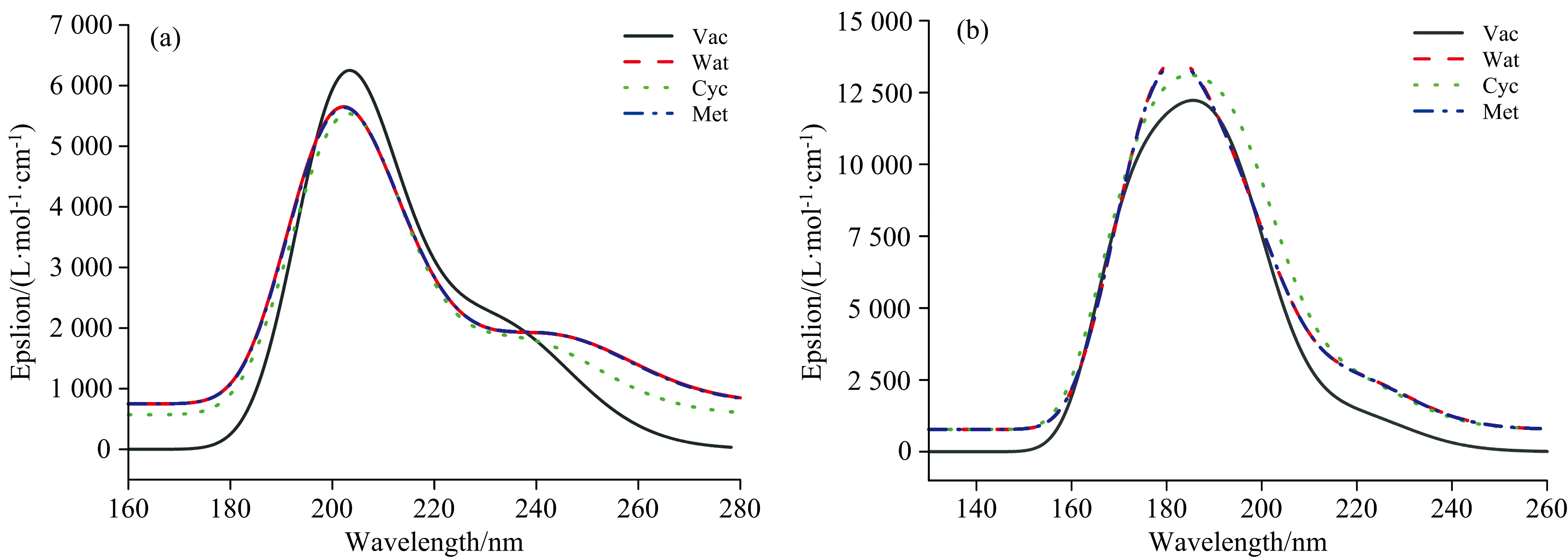 | 图4 B3LYP/6-311++G(2d, p) (a)和ω B97XD/6-311++G(2d, p) (b)水平计算四种环境化合物Euphorikanin A的UV-Vis吸收光谱Fig.4 The UV-Vis spectra of Euphorikanin A at the B3LYP/6-311++G(2d, p) (a) and ω B97XD/6-311++G(2d, p) (b) levels of theory in four environments |
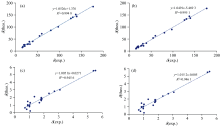
在结构优化的基础上, 采用规范独立原子轨道(GIAO)方法, 在B3LYP/6-311++G(2d, p)和ω B97XD/6-311++G(2d, p)水平计算了化合物Euphorikanin A的13C NMR和1H NMR化学位移。 表2为化合物Euphorikanin A在氯仿环境中13C NMR和1H NMR化学位移的理论数据。 对理论计算数据和实验数据进行线性回归, 得到描述理论化学位移和实验化学位移之间的关系[图5(a— d)]。 图5(a, b)两种方法计算得到的13C NMR理论数据与实验数据的相关性拟合方程分别为y=1.052 6x+1.376(R2=0.994 8), y=1.049 1x-5.469 3(R2=0.995 1)。 图5(c, d)1H NMR的理论数据与实验数据的拟合方程分别为y=1.005 8x+0.027 1(R2=0.945 6), y=1.015 2x-0.005(R2=0.946 1)。 (y是13C NMR和1H NMR的理论值, x是13C NMR和1H NMR的实验值, R2是理论值和实验值的相关系数)。 R2均大于0.94说明两种方法下得到的核磁数据理论结果与实际情况吻合度较高, 但ω B97XD方法计算得到的13C NMR数据与实验值更接近, R2大于0.99。
| 表2 B3LYP/6-311++G(2d, p)和ω B97XD/6-311++G(2d, p)水平计算化合物Euphorikanin A的13C NMR和1H NMR化学位移 Table 2 13C NMR and 1H NMR of the Euphorikanin A at the level of B3LYP/6-311++G(2d, p) and ω B97XD/6-311++G(2d, p) |
 | 图5 理论和实验13C NMR和1H NMR化学位移之间的关系 (a): B3LYP6-311++G(2d, p)计算的13C NMR; (b): ω B97XD/6-311++G(2d, p)计算的13C NMR; (c): B3LYP/6-311++G(2d, p)计算的1H NMR; (d): ω B97XD/6-311++G(2d, p) 1H NMRFig.5 Correlation graphs between experiments and calculated 13C NMR and 1H NMR chemical shifts (a): B3LYP6-311++G(2d, p) 13C NMR; (b): ω B97XD/6-311++G(2d, p) 13C NMR; (c): B3LYP/6-311++G(2d, p) 1H NMR; (d): ω B97XD/6-311++G(2d, p) 1H NMR |
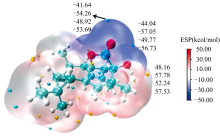
分子表面静电势(ESP)是通过识别亲核和亲电攻击的潜在位点来表征分子反应活性位点的一种方法。 ESP常被用于研究分子间的相互作用及分子识别, 解释分子化学反应活性与分子性质之间的关系[22]。 在B3LYP/6-311++G(2d, p)水平计算的四种环境中的化合物Euphorikanin A的分子表面静电势如图6所示, 蓝色区域表示负电荷区域, 易受到亲电试剂的攻击; 红色区域表示正电荷区域, 易受到亲核试剂的攻击; 白色表示零电位区域。 其中, 黄球对应静电势极大值点, 青球对应极小值点。 化合物Euphorikanin A的极大值点位于六元环的羟基附近, 化合物Euphorikanin A的极小值点位于内酯环的羰基附近, 说明化合物Euphorikanin A六元环的羟基可能为其活性位点。 随着环境溶剂极性增大, 化合物Euphorikanin A极值点增大, 活性增强。
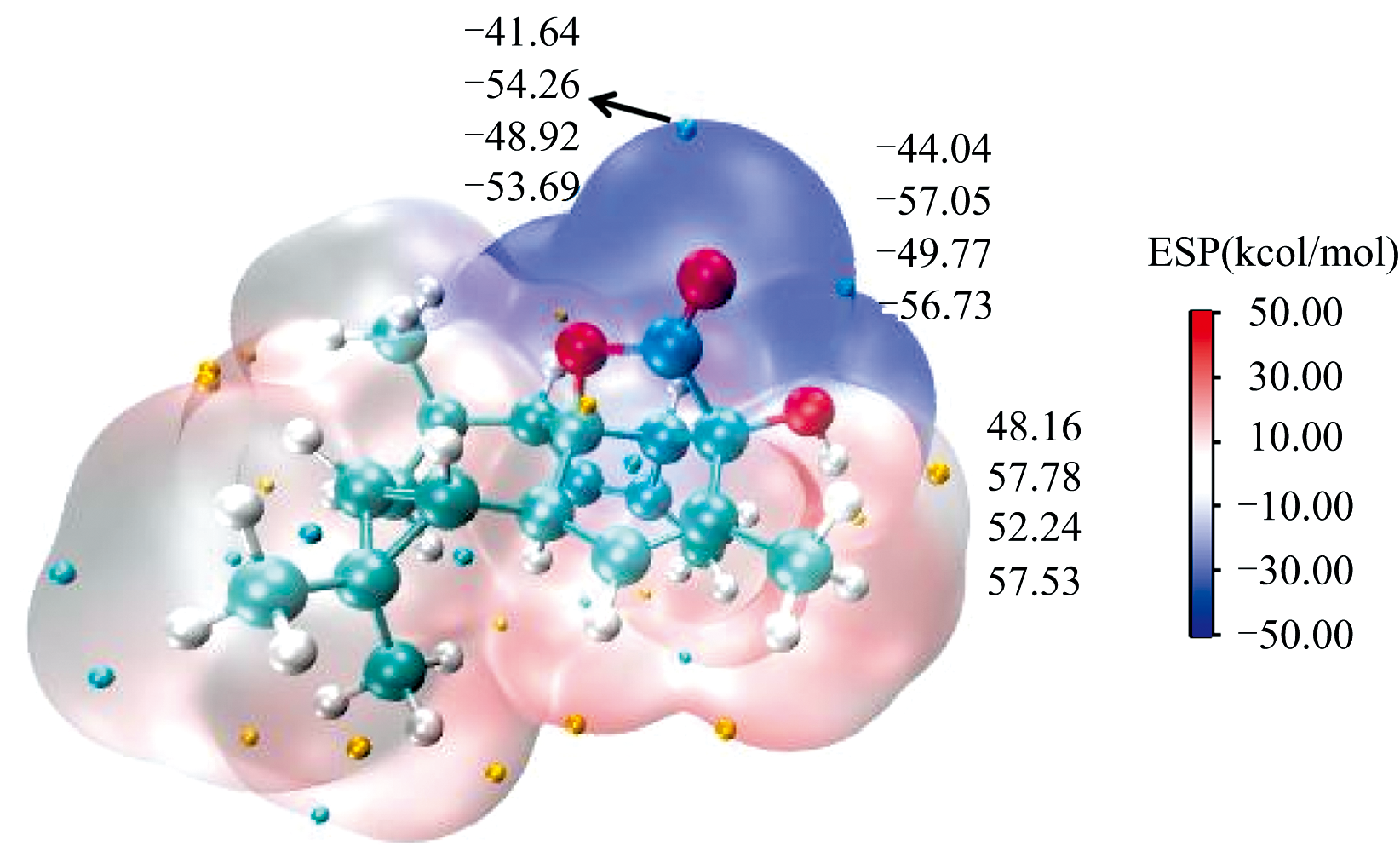 | 图6 B3LYP/6-311++G(2d, p)水平计算四种环境化合物Euphorikanin A的静电势Fig.6 Molecular electrostatic potential maps of of Euphorikanin A at the B3LYP/6-311++G(2d, p) levels of theory in four environments |
据文献公式计算了化合物Euphorikanin A的全局反应性参数, 结果见表3。 化学势μ 用于描述平衡构型的分子失去电子的能力, 其绝对值越大, 得到或失去电子的能力越强, 即反应性越强。 η 值表示微干扰下化学体系电子云抗畸变的能力, η 越小, 能力越弱。 全局亲电指数ω 用于描述体系得到电子时的稳定性, 其值越大, 亲电性越强, 反应活性强。 化合物Euphorikanin A的全局反应性参数与课题组前期计算的真空环境中镇痛药物吗啡的参数(μ =-0.14, η =0.10, ω =0.10)相似[23], 可能源于两者结构均为多环稠合结构, 构像限制也客观反映出化合物Euphorikanin A的成药可能性。
| 表3 B3LYP/6-311++G(2d, p)和ω B97XD/6-311++G(2d, p)水平计算四种环境Euphorikanin A的全局反应性描述符 Table 3 Global reactivity descriptors of Euphorikanin A at the B3LYP/6-311++G(2d, p) and ω B97XD/6-311++G(2d, p) levels of theory in four environments |
超过90%的候选药物由于出现安全性、 药效性和药代动力学等问题而无法上市。 在新药研发早期即开展候选药的成药性研究, 不仅有利于缩短药物的研发周期还能降低研发成本, 同时提高新药开发的成功率。 ACD/Percepta软件是基于QSAR(构效关系)模型预测化合物的吸收(A)、 分布(D)、 代谢(M)、 排泄(E)、 毒性(Tox)及理化性质(PhysChem)等方面的软件。 前期通过ACD/Percepta分析预测13种化合物的理化性质和药代动力学评价, 推测瑞德西韦可能对COVID-19疗效甚微或基本无效, 证实了软件对药物成药性和药代动力学预测的可靠性[24]。 化合物Euphorikanin A相关成药性评估参数和其药代动力学参数见表4, 其中Lipinski类药五规则是药物开发过程中常用的经验性规律, 类药性的筛选有利于获得药动学性质平衡的候选药, 化合物Euphorikanin A具有较好的类药性, 但溶解性较差。 药代动力学相关参数表明, 化合物Euphorikanin A对CYP450代谢酶1A2亚型不产生抑制作用。 值得注意的是, 化合物Euphorikanin A的Caco-2细胞膜通透性、 血脑屏障通过性和人肠道吸收性均表现出良好性能, 值得进一步实验研究。
| 表4 化合物Euphorikanin A的成药性评估 Table 4 Drug-like evaluation of Euphorikanin A |
在B3LYP/6-311++G(2d, p)和ω B97XD/6-311++G(2d, p)水平下, 计算分析了化合物Euphorikanin A的分子结构、 谱学性质和分子表面静电势, 结合概念密度泛函数预测反应活性位点, 采用ACD/Percepta软件预测其吸收、 分布、 代谢、 排泄/毒性(ADME/Tox)。 化合物Euphorikanin A具有唯一药效构像, 在不同溶剂环境, 化合物几何结构参数值相近, 计算值与晶体参数吻合较好。 计算红外光谱特征与实验吻合, 水环境中计算的IR光谱更接近真实环境, 理论计算的校正因子为0.94。 化合物Euphorikanin A紫外光谱最大吸收峰在193.88~201.75 nm。 两种方法得到的核磁数据理论结果与实际情况吻合度较高, R2均大于0.94。 化合物Euphorikanin A的极大值点位于六元环的甲基附近, Euphorikanin A的极小值点位于内酯环的羰基附近, 其六元环的羟基可能为其活性位点。 全局反应性参数与镇痛药物吗啡相似, 客观反映出化合物Euphorikanin A的成药可能性。 化合物Euphorikanin A的Caco-2细胞膜通透性、 血脑屏障通过性和人肠道吸收性在药代动力学预测中表现出良好性能。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|