





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函理论的蒽醌太赫兹吸收光谱模拟
[张同军 , 郝建军]
, 郝建军]
, 郝建军]
|
|


作者简介: 张同军, 1976年生,山东科技大学电子信息工程学院副教授 e-mail: supoptimal@163.com
蒽醌是一种平面结构的具有大环共扼体系的有机化合物, 在染料、 造纸、 生物和医药等众多行业有着广泛的应用。 为研究蒽醌在太赫兹频段的特征吸收和其分子晶体结构之间的关系, 基于密度泛函理论(DFT)并利用太赫兹时域光谱(THz-TDS)技术对蒽醌晶体的太赫兹吸收谱进行了理论模拟和实验研究。 首先利用THz-TDS系统测量了室温下蒽醌晶体在0.5~3.0 THz频段的特征吸收谱, 发现蒽醌晶体在该频段内有6个明显的特征吸收峰, 分别位于0.95, 1.05, 2.09, 2.25, 2.49和2.78 THz处。 为深入解析蒽醌在太赫兹频段实验特征谱的产生机理, 基于密度泛函理论分别对蒽醌单分子模型和晶胞模型进行了理论模拟计算。 使用Gaussian09软件对蒽醌单分子模型进行的理论计算, 采用了基于DFT的B3LYP杂化泛函方法和6-311G(d, p)基组, 几何结构优化和振动频率计算在相同水平上进行, 模拟计算结果与实验测量数据存在明显的差异, 说明单分子模拟存在一定的局限性。 使用MS8.0软件包中适合计算周期性结构的CASTEP模块对蒽醌晶胞模型进行理论计算, 采用了基于平面波赝势和广义梯度近似(GGA)的PBE、 PW91, WC, PBEsol和RPBE五种交换相关泛函, 几何结构优化和晶格动力学计算在相同水平上进行。 将蒽醌单分子和晶胞的理论模拟结构参数(键长、 键角)分别与其X射线衍射实验测量的结构参数进行了详细的比较分析, 发现基于PBE方法获得的固态仿真结果中分子的结构参数与X射线衍射实验数据的一致性最好。 同时用PBE和RPBE方法获得的理论仿真谱与实验吸收谱比较吻合。 因此, 基于PBE和RPBE的计算结果对实验特征吸收峰进行了振动模式指认。 研究结果表明, 蒽醌晶体在太赫兹频段的特征吸收主要来源于晶体中由C—H…O分子内氢键主导的蒽醌环和苯环基团的整体振动以及由分子间弱相互作用引发的集体振动模式。
Anthraquinone is an organic compound with a planar structure and a macrocyclic conjugated system, which has a wide range of applications in various industries such as dyes, papermaking, biology, and medicine. To investigate the relationship between the characteristic absorption of anthraquinone in the terahertz region and its molecular crystal structure, theoretical simulation and experimental research of terahertz absorption spectrum was carried out by using density functional theory (DFT) and terahertz time-domain spectroscopy (THz-TDS). Firstly, the characteristic absorption spectrum of anthraquinone in the frequency range of 0.5~3.0 THz at room temperature was measured using the THz-TDS system. It was found that anthraquinone crystals have six distinct characteristic absorption peaks in this frequency range, located at 0.95, 1.05, 2.09, 2.25, 2.49, and 2.78 THz, respectively. For deeply analyzing the generation mechanism of the characteristic spectra of anthraquinone in the terahertz frequency band, theoretical simulation calculations were conducted on the single molecule model and the cell model of anthraquinone based on density functional theory. Theoretical calculations were conducted on the anthraquinone single molecule model using Gaussian09 software-based DFT theory with the B3LYP hybrid functional method and 6-311G (d, p) basis set. Geometric structure optimization and vibration frequency calculation were carried out at the same level. The simulation results showed significant differences from experimental measurement data, indicating that single-molecule simulation has certain limitations. Theoretical calculations were carried out on the anthraquinone crystal cell model using the CASTEP module, which is suitable for calculating periodic structures in the MS 8.0 software package. Five exchange-related functionals, PBE, PW91, WC, PBEsol, and RPBE, based on plane wave pseudo potential and generalized gradient approximation (GGA), were used. Geometric structure optimization and lattice dynamics calculations were performed at the same level. A detailed comparative analysis was conducted between the simulated structural parameters (bond length, bond angle) of anthraquinone single molecules and crystal cells and the structural parameters measured by X-ray diffraction experiments. It was found that the consistency between the molecular structural parameters and X-ray diffraction experimental data was the best in the solid-state simulation results obtained based on the PBE method. The theoretical simulation spectra obtained by PBE and RPBE methods agree with the experimental absorption spectra. Therefore, the vibration mode assignment of the experimental characteristic absorption peak was carried out based on PBE and RPBE calculation results. The study indicates that the characteristic absorption of anthraquinone crystals primarily originated from the overall vibration of the anthraquinone ring and benzene ring groups dominated by C—H…O intermolecular hydrogen bonds in the crystal, as well as the collective vibration mode caused by weak intermolecular interactions.
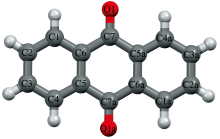
蒽醌是蒽的衍生物, 又名9, 10-蒽二酮, 分子式为C14H8O2, 其分子结构如图1所示。 蒽醌类化合物具有平面结构的大环共扼体系[1], 与金属离子配位能形成各种颜色的络合物, 在染料工业上有重要地位。 蒽醌作为燃料中间体可用于生产蒽醌系分散染料、 反应染料、 还原染料和酸性染料等四百多个品种的合成染料。 蒽醌作为蒸煮剂可用于造纸制浆行业, 作为脱硫剂可用于农药化肥行业, 作为光敏剂可用于光盘信息存储行业。 蒽醌类化合物还有重要的药用价值, 比如大黄、 芦荟的药理作用和有效成分即为蒽醌衍生物, 米托蒽醌还是一种被广泛使用的抗肿瘤药物。 作为蒽醌类化合物的基本母核, 蒽醌本体上常以羧基、 甲基、 甲氧基和羟基等为取代基进而形成种类繁多的蒽醌衍生物[2]。 通过紫外光谱(UV)、 圆二色光谱(CD)和荧光光谱等光谱学方法的研究发现, 许多蒽醌衍生物都和细胞的功能有关, 还和体内的电子及氢原子转移的机理有关, 某些蒽醌衍生物还可以和DNA相互作用, 在特定位置裂解DNA的单链或双链。 当前, 对蒽醌分子及晶体内部结构的太赫兹光谱研究还处于起步阶段。
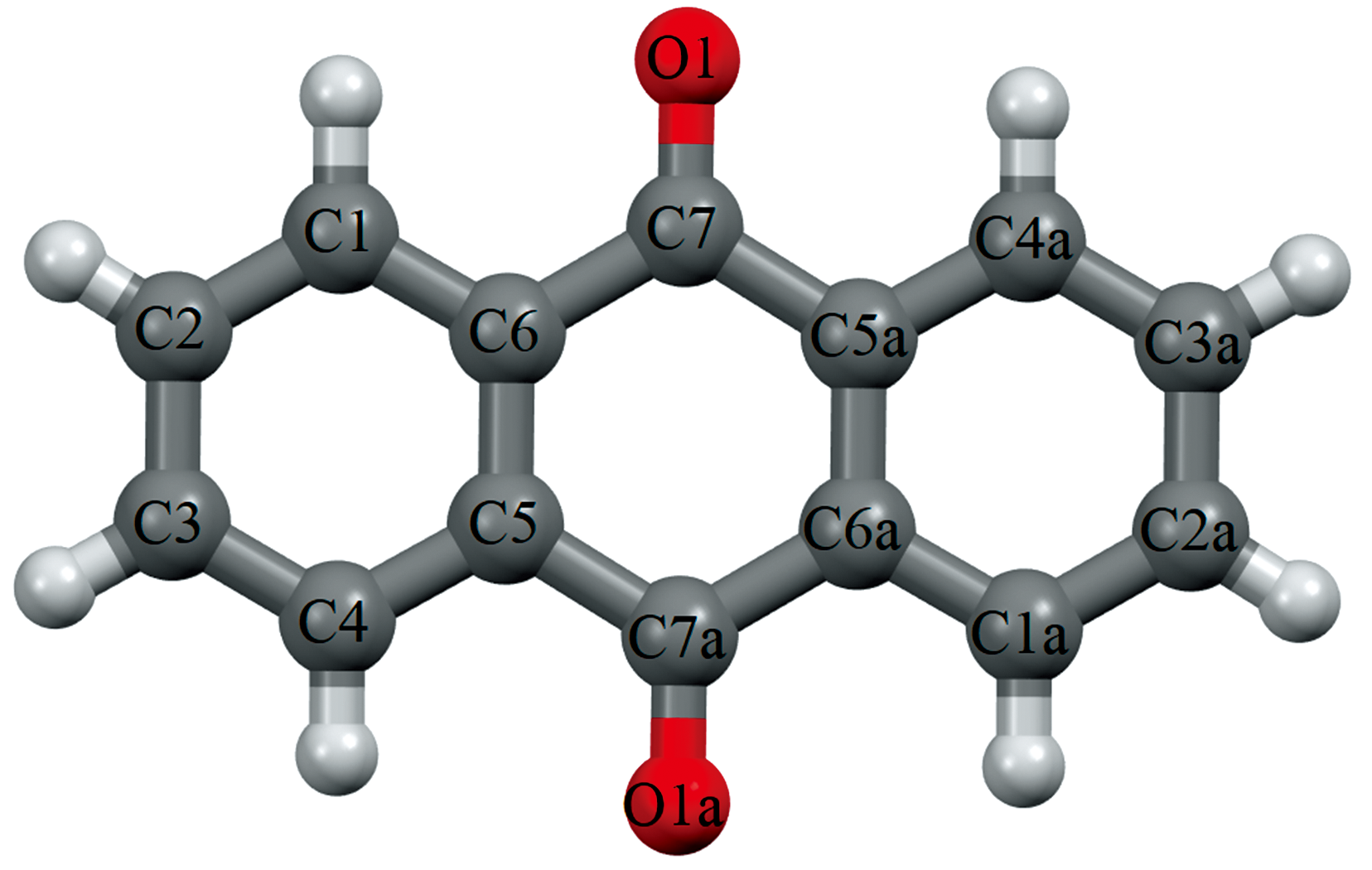 | 图1 带标号的蒽醌分子的结构图Fig.1 The labeled molecular structure of Anthraquinone |
位于电磁波谱远红外频段的太赫兹(THz)波, 承载着有机物质与电磁辐射之间复杂的物理、 化学相互作用。 这个波段的光子能量在1~100 meV这样一个相当宽泛的范围内, 不仅不会对物质产生有害的光致电离, 而且由于许多分子体系的能级跃迁也处在这个量级, 因而会与THz波发生强烈的相互作用。 近年来, 太赫兹时域光谱(Terahertz time-domain spectroscopy, THz-TDS)技术已经迅速发展成为一种可以应用于有机分子检测与研究的新型光谱技术, 许多有机分子晶体通过THz-TDS实验获得了具有指纹性特征的吸收峰。 有机物晶体的偶极和色散相互作用、 分子间氢键、 范德华力等弱相互作用, 晶格的声子振动、 骨架振动以及分子内的低频集体振动模式等正好出现在THz频段内, 使得利用THz-TDS技术研究蒽醌等有机物的特征吸收、 分子结构和晶体结构成为可能, 吸引了国内外众多研究者的兴趣[3]。 与实验同时, 许多学者开始借助量子化学计算对目标物质的太赫兹特征吸收谱进行理论模拟分析[4]。 密度泛函理论(density functional theory, DFT)是一种基于Hohenberg-Kohn定理的多电子体系结构的计算化学研究方法。 DFT可对孤立分子和分子晶体的振动模式进行理论计算模拟和光谱解析, 进而确定物质的特征吸收峰的形成机理和振动模式归属[5]。 到目前为止, 对于蒽醌的太赫兹光谱研究大多专注于实验研究和定量测试[6], 或以单分子为模型进行理论计算模拟[7]。
本工作中利用THz-TDS技术获得了室温条件下蒽醌在0.5~3.0 THz范围内的特征吸收谱, 并基于密度泛函理论分别对蒽醌的孤立分子和晶胞进行化学建模、 几何结构优化和晶格动力学计算。 该研究对于揭示蒽醌在太赫兹波段特征吸收峰的形成机制, 获得其分子结构与太赫兹特征吸收谱之间的关系等, 具有重要的理论价值和实际意义。
实验所用的蒽醌样品为浅黄色结晶针状固体, 购于上海麦克林生化科技股份有限公司, 纯度大于99%, 样品制备之前未做深度结晶提纯。 将蒽醌样品与高纯度聚四氟乙烯粉末以1∶ 10的比例混合后放入研钵内充分研磨, 用精密电子天平称取150 mg均匀混合物放入FW4A型压片机以12 MPa的压力压成直径13 mm, 厚度1.1 mm的圆形薄片。 样品片结构均匀, 前后表面光滑且平行。
THz光谱测量所使用的实验装置是由德国BATOP公司生产的TDS1008系统[8]。 系统配备了Spectra Physics公司生产的MaiTai锁模钛蓝宝石飞秒激光器, 激光中心波长780 nm, 重复频率80 MHz, 平均功率1.5 W。 系统通过泵浦激光束激发光电导天线产生THz脉冲, 探测光束通过自由空间电光采样的方法对包含样品信息的THz脉冲进行探测, 可实现高达2.0 GHz频谱分辨率和0.05~4.0 THz的光谱范围。 实验测量过程中, 需要将干燥的氮气持续吹扫到样品室中, 以使空气中水蒸汽的影响降至最低, 保持实验环境相对湿度低于3%。
实验数据的获取是在室温295 K, 实验箱充满氮气的环境下进行的。 图2(a)所示为THz波穿过蒽醌和氮气环境所获得的样品信号和参考信号的时域光谱。 由图中可以看出, 相比于参考信号, 样品信号有时间上的延迟和幅度上的收缩, 这是由于THz波穿过样品时折射率变大, 且样品对THz波的吸收所致。 对图2(a)的时域信号进行快速傅里叶变换后可以得到样品和参考信号的频谱图, 如图2(b)所示。 根据Timothy和Duvillaret等[9, 10]提出的提取材料THz波光学参数的物理模型对实验数据进行处理。 通过图2(a)可以看出, 时域光谱同时获取了信号的振幅和相位信息, 因此可以通过计算得到样品的折射率和吸收系数。
 | 图2 蒽醌和参考(氮气)的时域谱(a)和频域谱(b)Fig.2 Time-domain spectrum (a) and frequency-domain spectrum (b) of Anthraquinone and the Reference (nitrogen) |
图3所示为蒽醌样品在0.5~3.0 THz范围内的实验吸收谱。 由图可见, 蒽醌存在6个明显的特征吸收峰, 分别位于0.95, 1.05, 2.09, 2.25, 2.49和2.78 THz处, 最强的特征吸收峰位于2.25 THz处, 其左右两边各出现了一个肩峰, 另外的三个吸收峰为弱吸收峰。 吸收谱的基线整体上随频率的升高呈缓慢上倾趋势, 这可能是由于光散射和样品宽而无结构的吸收所致。
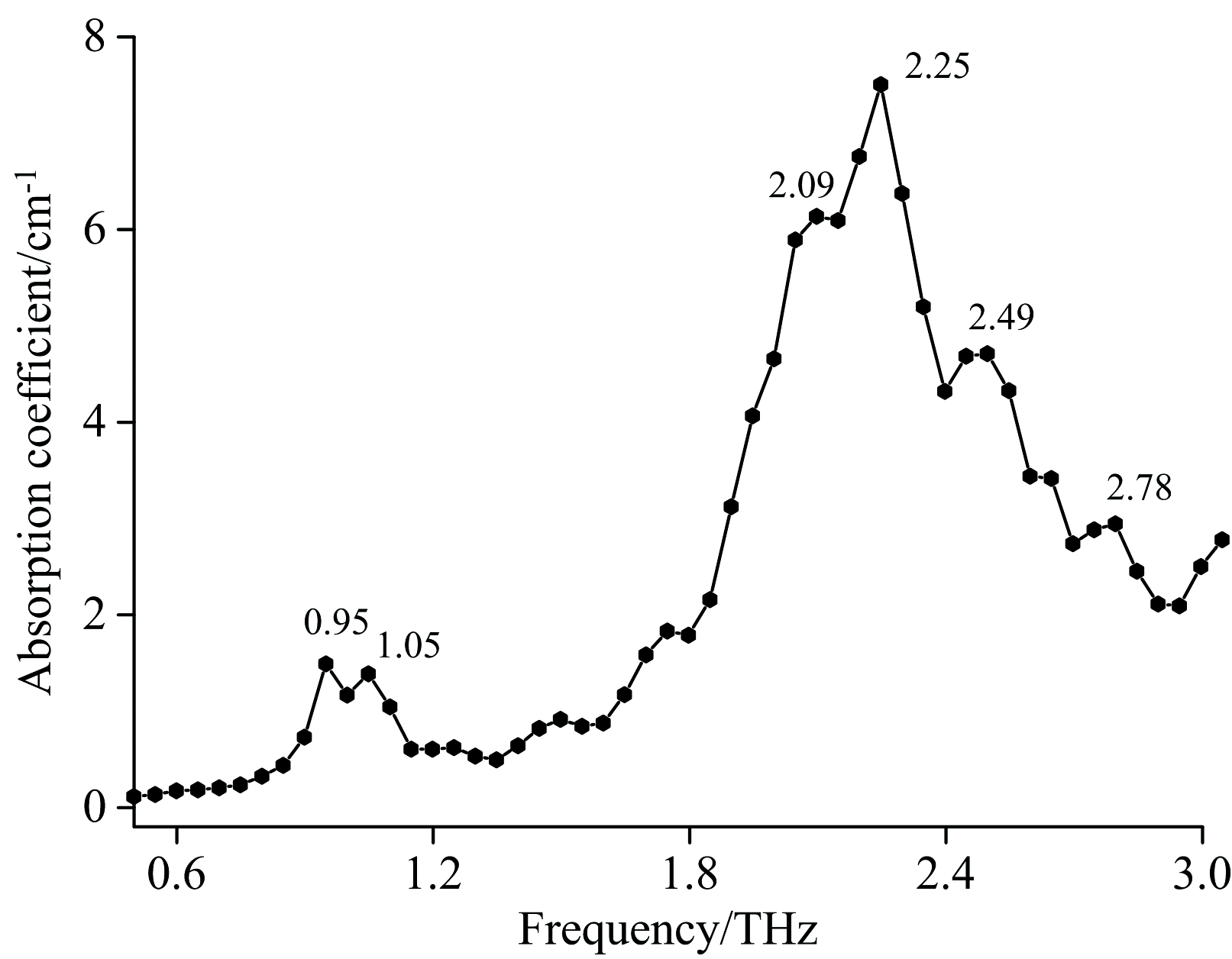 | 图3 蒽醌的THz实验吸收谱Fig.3 The THz experimental spectrum of Anthraquinone |
为准确解析蒽醌太赫兹实验特征吸收谱的产生机理, 使用基于密度泛函理论的量子化学计算方法, 并借助Gaussian软件包对孤立单分子模型、 Materials Studio软件包对晶胞模型进行了几何优化, 能量和振动频率计算。
气态孤立分子的理论计算模型的建模流程如下: 使用Chemdraw软件画出蒽醌单分子的化学结构式, 然后导入Chem3D软件中构建分子的立体结构并进行构象优化, 最后在GaussView软件中完成高斯计算输入文件的构建。 孤立分子模型的模拟计算在Gaussian09程序包中进行, 采用基于DFT的B3LYP杂化泛函方法, 选取6-311G(d, p)基组进行几何优化, 获得其基态稳定构型作为初始构型, 并在相同水平上进行了振动频率计算。
为探究蒽醌晶胞中分子内氢键、 分子间偶极和色散相互作用力、 范德华力等分子间弱相互作用对太赫兹特征吸收谱的影响, 对蒽醌的晶胞模型进行了仿真计算。 蒽醌的晶胞模型来自剑桥晶体结构数据库(Cambridge Crystallographic Data Centre, CCDC), 其CSD参考代码为ANTQUO13。 该晶胞结构是由Fu[11]等在162 K的温度下通过X射线衍射技术获得的。 如图4所示, 蒽醌的晶胞属于单斜晶系, 每个晶胞中包含有两个蒽醌分子, 空间群为P21/c; 晶格尺寸为: a=7.867 Å , b=3.895 Å , c=15.651 Å , V=468.13 Å 3; 三条晶轴之间的关系为: α = 90° , β =102.55° , γ =90° 。
 | 图4 蒽醌的晶胞结构Fig.4 The unit cell structure of Anthraquinone |
晶胞结构的理论模拟计算是在量子化学计算软件包Materials Studio8.0的CASTEP[12]模块中进行的, 该模块特别适合计算周期性结构。 采用基于平面波赝势的密度泛函理论(DFT)和广义梯度近似(GGA)方法的PBE、 PW91, RPBE, PBEsol和WC五种交换相关泛函对蒽醌晶胞模型进行几何结构优化和振动频率计算。 几何优化时的设置采用了BFGS拟牛顿算法中的线搜索(line search)方法, 在对晶胞整体进行优化的同时还对蒽醌的原子位置进行了优化。 几何优化时的收敛精度设置为“ fine” , 相应的四个主要参数的收敛阈值设置: 最大能量(Energy) 10-5 eV· atom-1, 最大受力 (Max.force) 0.03 eV· Å -1, 最大压力(Max.stress) 0.05 GPa, 最大位移 (Max.displacement) 10-3 Å 。 平面波截断能设置为103 eV, 选择模守恒赝势基组, 使用Γ 点计算振动频率, 在2× 3× 1 的Monkhorst-Pack栅极上进行电子态的布里渊区采样。
表1和表2分别列出了蒽醌分子通过X射线衍射 (XRD) 实验获得的键长、 键角的结构参数, 同时列出了孤立分子和晶胞模型经过几何优化后的相应结构参数, 并且给出了各理论模拟值与实验测量值之间的均方根偏差RMSD[13]。 RMSD值可以直观地表示出各种理论泛函方法模拟计算的键长、 键角参数与XRD实验测量值之间的偏离程度, 偏离程度越小说明该理论方法的模拟结果越接近分子的实际情况。 RMSD的计算公式如式(1)所示
式(1)中, xcal和xexp分别是蒽醌分子结构参数的理论计算值和实验测量值。
| 表1 蒽醌分子的六种理论键长值(Å )及其相对于实验测量数据的RMSD值 Table 1 Six calculated bond lengths (Å ) of Anthraquinone molecule and their RMSD values compared to experimental data |
| 表2 蒽醌分子的六种理论键角值(° )及其相对于实验测量数据的RMSD值 Table 2 Six calculated bond angles (° ) of Anthraquinone molecule and their RMSD values compared to experimental data |
通观表1数据可知, 蒽醌晶胞的键长模拟值与其实验值的平均绝对偏差大都在0.001 5 nm之内。 相比之下, 孤立分子的键长模拟值与实验值之间的差值则非常之大, 最高达到了0.014 nm, 是晶胞模型相应参数的十多倍。 由表2数据可知, 蒽醌晶胞的键角的模拟值与其实验值的平均绝对偏差大都在0.8° 之内, 而孤立分子的对应差值则达到了11.46° 。 可见孤立蒽醌分子在不受晶格约束的情况下, 其结构参数具有非常大的弹性。
图5(a)、 (b)分别给出了蒽醌晶胞的分子内键长、 键角的理论计算值与实验测量值之间偏差的变化图, 更直观地展示了蒽醌分子的五种理论模拟结构参数与实验结构参数之间的差异。 由于基于DFT的B3LYP方法计算出的孤立分子的结构数据与实验数据的偏差太大, 没有在离差图中体现。 基于B3LYP方法的单分子键长的RMSD值为0.111 5, 相较于其他五种基于固态GGA泛函方法的晶胞中分子的键长RMSD值, 增大了一个数量级, 说明蒽醌在太赫兹频段基于单分子的结构模拟存在较大偏差。
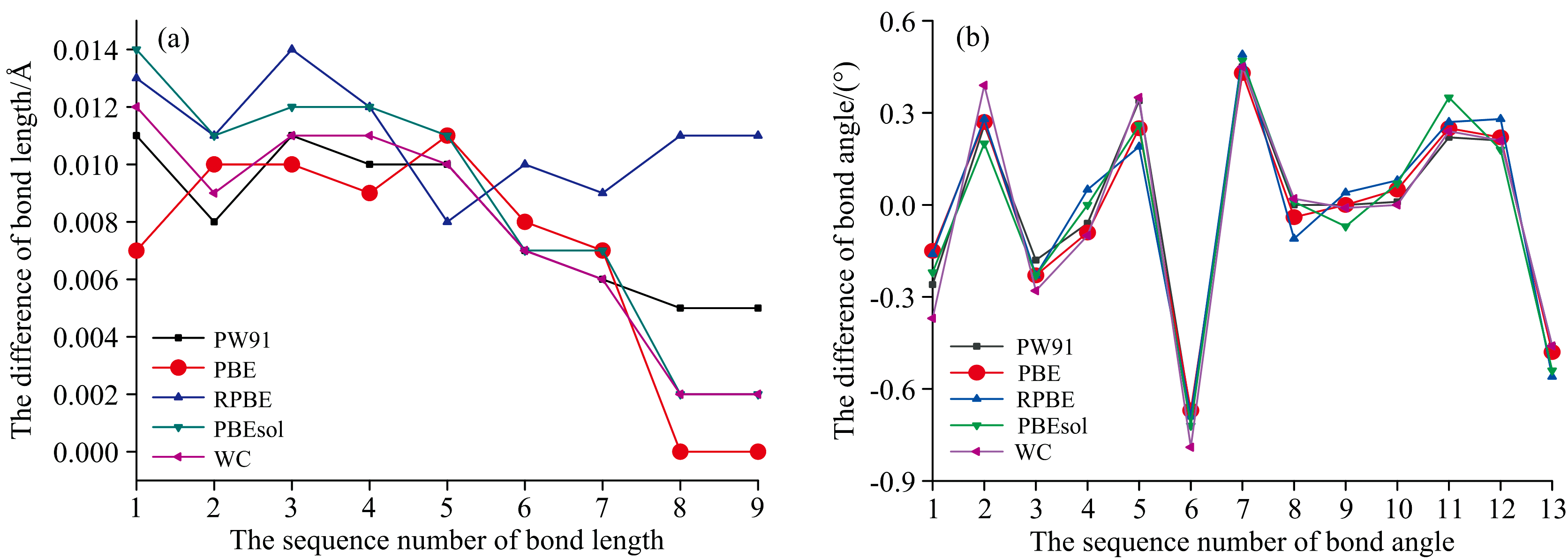 | 图5 蒽醌分子的键长(a)、 键角(b)的差值变化图Fig.5 Bond length (a) and bond angle (b) differences between simulations and experimental data |
由图5(a)可以看出, 基于PBE泛函的计算结果与实验值之间的差值大都在0.001 nm之内, 且其键长RMSD值也是所有模拟计算结果中最小的, 仅为0.007 9, 说明这种计算水平下得到的模拟结果相当可靠。 蒽醌晶胞分子中苯环的六个C— C键的键长在1.384~1.406 Å 之间, 介于1.339 Å 的C— C单键和1.540 Å 的C=C双键之间, 呈现出明显的共轭特征。 分子中C— O键的键长为1.221 Å , 小于标准的C— O单键的键长1.430 Å , 说明也参与了分子的整体共轭, 同时验证了蒽醌分子的内部形成了大π 共轭体系。
由表2还可以看出, 蒽醌晶胞的键角模拟值与其实验值的平均绝对偏差全部都在1° 之内, WC泛函计算结果出现的最大偏差也仅有0.79° , 说明基于固态泛函的晶胞模拟能很好地实现对实验结果的仿真。 由图5(b)可以看出, 五种固态GGA泛函的键角模拟值与实验值偏差的变化趋势表现出了很好的一致性。 相比之下, 基于B3LYP的单分子键角模拟结果表现出了与实验数据的明显差别, 最大偏差达到了11.46° , 相应的键角RMSD值达7.981, 是五种固态GGA算法模拟结果的十倍以上。 这种差异说明基于单分子模型的仿真结果与实验测量值存在较大差异, 因此单纯用单分子的计算谱来模拟太赫兹频段的实验谱, 必定是存在较大的偏差。 PBE泛函的仿真结果与实验值之间的整体偏差最小, RMSD值也最小, 仅为0.303 6, 说明用PBE固态泛函的理论计算谱来仿真实验谱更好。
图6给出了蒽醌的标准摩尔实验吸收谱与基于B3LYP方法的孤立分子模型、 基于五种GGA泛函的晶胞模型的模拟仿真谱的对比。 图6中的模拟谱是利用开源软件Multiwfn3.0[14]绘制的, 以洛伦兹函数作为展宽函数并设定4 cm-1的半高宽(FWHM)值对相应谐振频率处红外振动强度进行拟合得到。
 | 图6 蒽醌的标准摩尔实验吸收谱与其模拟仿真谱的对比Fig.6 The standard molar absorption spectrum of Anthraquinone compared to its simulation spectra |
基于DFT理论、 B3LYP泛函和6-311G(d, p)基组在Gaussian09软件包中对蒽醌孤立分子进行的计算在0.5~3.0 THz范围内仅产生了1个简正振动模式, 位于2.82 THz处。 该简正模式与实验谱中2.78 THz处的特征吸收峰相吻合, 而所有基于固态泛函的晶胞模拟都没有在此处产生特征吸收, 所以认为蒽醌在2.78 THz处的吸收峰来源于分子内振动模式, 在Gaussview软件的振动分析中显示为两个对称C=O键的面外摇摆。 蒽醌分子包含24个原子, 有66个基频振动, 按D2h对称性可将其振动方式分类为12Ag+4B1g+6B2g+11B3g+5Au+11B1u+11B2u+6B3u, 其中Ag, B1g, B2g, B3g表现为拉曼活性, B1u, B2u, B3u表现为红外活性, Au既表现拉曼活性又表现红外活性。 2.78 THz处的特征吸收峰是实验谱中唯一表现为B2u红外活性的分子内振动模式, 其他的特征吸收都需要由晶胞模型进行解释。
为更准确全面地解释蒽醌的太赫兹实验吸收谱, 并为了验证GGA密度泛函模拟蒽醌晶体的性能, 利用PBE、 PW91、 RPBE、 PBEsol和WC五种泛函对蒽醌的晶胞模型进行了几何优化和晶格动力学计算, 重复执行计算直到结果中无虚频出现, 证明几何优化得到的结构是一个稳定的形态。 由图6可知, 在0.5~3.0 THz范围内, PBE、 RPBE和WC泛函各产生了4个简正模式, PW91和PBEsol泛函分别产生了3个和5个简正模式。 通过将这些简正模式的频率位置和振动强度与实验吸收峰进行对比, 可以实现对蒽醌晶体太赫兹实验吸收谱的振动模式的指认。 由图6可知, 任何单一泛函的计算结果都不能实现对实验吸收谱的完美解释。 PBE泛函从理论上改进了PW91, 能精确地描述原子、 分子、 晶体的自旋局域密度, 进而可以更精确地描述体系的能量, 使其成为泛函中的主流。 PBE和RPBE泛函的模拟结果与实验吸收谱在频率位置、 振动强度以及谱线形状上都比较接近, 这可能是因为两者优化后的结构参数比其余三种泛函的优化结果更接近于晶体本身。 特别是PBE, 其优化后的结构参数无论键长还是键角的RMSD值都是五种泛函中最小的。 尽管WC和PBEsol分别从表面吸附和调整泛函中自相互作用误差等方面对PBE进行了改进, 但从模拟结果来看并不适合对蒽醌太赫兹实验吸收谱的解析。 同时使用PBE和RPBE可以更好地解析蒽醌实验吸收谱并实现振动模式的指认。
由前述分析可知, PBE和RPBE泛函的模拟结果与实验吸收谱在频率位置、 振动强度以及谱线形状上最为接近。 因此, 用PBE和RPBE的简正模式对相应的实验特征峰进行振动模式指认更为准确。
表3列出了蒽醌在0.5~3.0 THz范围内的实验吸收峰及其对应的PBE、 RPBE以及B3LYP模拟吸收峰的频率位置, 并给出了振动模式归属。 分子晶体的振动模式分为分子间振动和分子内振动, 分子间振动反映了晶体的结构及其对称性, 分子内振动主要是指分子内的变形振动。 分子间振动的频率通常低于分子内振动的频率。 PBE和RPBE理论预测的蒽醌晶体的所有振动模式都属于分子间振动模式, 而B3LYP指认的振动属于分子内振动。
| 表3 蒽醌吸收峰振动模式归属 Table 3 Vibrational modes Assignment of Anthraquinone's absorption peaks |
图7给出了前四个实验吸收峰对应的简正振动模式的原子位移矢量图, 其中0.86和0.96 THz是基于RPBE泛函的, 2.03和2.35 THz是基于PBE泛函的。 这些振动模式直观地展示了蒽醌晶体内复杂的振动现象, 说明蒽醌在THz频段的特征吸收主要来自以C— H…O分子内氢键主导的蒽醌环和苯环基团的整体振动, 以及由晶体内的分子间弱相互作用带动的骨架振动和羰基基团的面外弯曲振动。
 | 图7 蒽醌晶胞的基于PBE和RPBE泛函的简正振动模式Fig.7 The simulated displacement vectors of anthraquinone crystal cell based on PBE and RPBE functionals |
利用太赫兹时域光谱系统获取了蒽醌晶体在0.5~3.0 THz频率范围内的时域谱、 频域谱和吸收谱, 实验及数据处理结果显示蒽醌在该频段内具有六个特征吸收峰。 分别采用基于DFT理论的BELYP/6-311G(d, p)方法和多种GGA泛函对孤立蒽醌分子和蒽醌晶胞的初始构型进行了几何结构优化和振动频率计算。 从分子结构和吸收谱峰位两方面对实验测量和理论模拟的数据进行了详细的比较分析, 发现基于晶胞模型的GGA泛函模拟结果与实验结果的吻合度更高。 说明氢键、 分子间偶极色散、 范德华力和晶格振动等分子间弱相互作用对蒽醌在太赫兹频段的吸收有重要影响。 可见对于蒽醌晶体样品在太赫兹频段的理论模拟, 应尽量选取能体现分子间作用的晶胞模型作为计算的初始构型。 基于晶胞模型的五种GGA泛函对蒽醌分子结构数据的模拟效果较好, 也比较一致。 选取与实验谱峰位最为接近的PBE、 RPBE泛函的计算结果对太赫兹实验吸收峰对应的振动模式进行了归属。 研究表明, 蒽醌晶体在THz频段的特征吸收主要来源于由C— H…O分子内氢键主导的蒽醌环和苯环基团的整体振动以及由分子间弱相互作用引发的集体振动模式。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|