

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
煤系石墨的微观结构特征和热膨胀行为
[李焕同1  , 张谦
, 张谦1 , 邹晓艳2, * , 张卫国1 , 林柯瑾1 ]
, 张谦, 张卫国|
|


作者简介: 李焕同, 1986年生, 西安科技大学地质与环境学院副教授 e-mail: htlcumt@126.com
石墨是一种由sp2杂化碳原子组成的层状结构碳材料, 煤系石墨是由煤经高温变质形成的隐晶质石墨, 其微观结构具有显著的各向异性。 选取了无烟煤-煤系石墨序列的4个样品, 通过X射线衍射(XRD)、 高分辨透射电镜(HRTEM)、 X射线光电子能谱(XPS)、 拉曼光谱(Raman)、 傅里叶红外光谱(FTIR)和紫外-可见光-近红外漫反射测试(UV-Vis-NIR)等手段, 分析其微观结构特征及热膨胀行为, 并利用高温原位XRD分析二者之间的定量关系。 结果表明: 随着石墨化程度的增强, 煤系石墨微晶尺寸和沿 c轴方向的堆砌高度显著增大, 各向异性特征更加明显。 不同类型缺陷对应的 ID1/ ID2比值不同, 基面的 ID1/ ID2高于边缘面。 HRTEM显示高变质无烟煤的芳香层片具有局部定向域, 堆叠层数较少且延伸长度不稳定, 微晶表面或边缘存在无定形碳。 含氧官能团主要集中在石墨微晶的缺陷区域, 随着石墨化度的提高, 氧元素含量降低, 羰基C═O比例减少。 煤系石墨中多环芳烃的存在使π—π*跃迁吸收峰蓝移至208 nm附近, 仿真颜色分布于Cool Gray 9 CP至2 CP, 石墨化程度越高, 反光能力越强。 热膨胀行为方面, 石墨的热膨胀系数表现出明显的各向异性, 平行于基平面方向的热膨胀系数小于垂直方向。 随着温度升高, 石墨微晶在 c轴方向的膨胀由层间距的等效膨胀调节, 而在 a轴方向, 晶格缺陷的形成导致平均微晶尺寸(W-Hsize)降低。 石墨化程度越高, 微观结构越完善, 各向异性越显著, 热膨胀行为也表现出明显的各向异性。
Graphite is a layered carbon material composed of sp2 hybridized carbon atoms. Coal-measure graphite, a cryptocrystalline form of graphite derived from coal through high-temperature metamorphism, exhibits significant anisotropy. In this study, four samples from the anthracite-coal measure graphite series were selected and analyzed for their microstructural characteristics and thermal expansion behavior using X-ray diffraction (XRD), high-resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, Fourier transform infrared spectroscopy (FTIR), and ultraviolet-visible-near-infrared diffuse reflectance testing (UV-Vis-NIR). The quantitative relationship between these properties was also investigated using in situ high-temperature XRD. The results show that as the graphitization degree increases, the microcrystalline size and stacking height along the c-axis direction of coal-measure graphite significantly increase, and the anisotropic characteristics become more pronounced. The ID1/ ID2 ratio, which corresponds to different defect types, varies, with the basal plane exhibiting a higher ID1/ ID2 ratio than the edge plane. HRTEM reveals that the aromatic layers of highly metamorphosed anthracite exhibit local orientation domains, with fewer stacking layers and unstable extension lengths, and that amorphous carbon is present on the microcrystalline surface or edges. Oxygen-containing functional groups are mainly concentrated in the defective regions of graphite microcrystals. As the graphitization degree increases, the oxygen content decreases, and the proportion of carbonyl groups (C═O) decreases. The presence of polycyclic aromatic hydrocarbons in coal-measure graphite -shifts the π—π* transition absorption peak to approximately 208 nm, with a simulated color distribution ranging from Cool Gray 9 CP to 2 CP. Higher graphitization degrees are associated with stronger reflectivity. Regarding thermal expansion behavior, the thermal expansion coefficient of graphite shows significant anisotropy, with the coefficient parallel to the basal plane being lower than that perpendicular to it. As the temperature rises, the expansion of graphite microcrystals along the c-axis is regulated by the corresponding increase in-the interlayer spacing. In contrast, in the a-axis direction, the formation of lattice defects decreases the average microcrystalline size (W-Hsize). The higher the graphitization degree, the more complete the microstructure, the more pronounced the anisotropy, and the more significant the anisotropic thermal expansion behavior.
石墨是一种层状结构的碳材料, 由sp2杂化碳原子组成六角形平面网状结构, 层间通过范德华力相互作用。 煤系石墨是指由煤经过高温变质作用形成的隐晶质石墨[1], 属于石墨的一种类型。 煤系石墨由多层石墨烯片堆叠而成, 在构造应力和温度作用下, 石墨微晶会发生择优取向生长, 沿c轴方向堆砌高度Lc增加, 当Lc≥ 30 nm时, 煤系石墨结构较为完善, 呈现显著的各向异性[2]。 煤系石墨的显微组分(如变镜质体)在反射单偏光下表现出多色性和强光学各向异性, 不同显微亚组分之间光性差异明显[3]; 在正交偏光加石膏试板观测条件下, 煤系石墨的石墨微晶呈一级黄和二级蓝干涉色, 指示石墨微晶不同的排列方向。 由于石墨微晶的择优取向, 煤系石墨的导热性在石墨层的平面方向上较强, 而在垂直方向上较弱。 石墨的热膨胀特性与其六方结构中平行和垂直于基面的晶格振动变化, 以及随温度变化的相关性质紧密相关(Riley理论)[4]。 深入研究煤系石墨的微观结构特征及其与热膨胀行为之间的内在联系, 对于优化其加工工艺、 拓展应用范围以及开发高性能石墨材料具有重要的理论和实际意义。
本工作在前期煤系石墨微晶结构各向异性和非均质性研究的基础上, 利用XRD、 HRTEM、 XPS、 Raman、 FTIR和UV-Vis-NIR等手段分析煤系石墨的微观结构特征, 并利用高温原位XRD分析了微观结构与热膨胀行为之间的定量关系。
样品采自湖南寒婆坳矿区稠木煤矿、 胜利煤矿、 稗冲煤矿和石巷里煤矿的井下煤层样, 编号分别为CM130N、 SL150S、 BC210和SXL130。 依据GB/T 212— 2008《煤的工业分析方法》、 GB/T 31391— 2015《煤的元素分析》以及GB/T 6948— 2008《煤的镜质体反射率显微镜测定方法》测试, 结果见表1。
| 表1 镜质体反射率、 工业分析和元素分析结果 Table 1 Vitrinite reflectance, proximate and ultimate analysis of coal measures graphite |
1.2.1 X射线衍射分析(XRD)
取脱矿煤粉(CM130N、 BC210、 SL150S和SXL130)在MSAL-XD2 X射线衍射仪上进行测试。 实验条件: Cu靶, K辐射, 管压36 kV, 管流30 mA, 发散狭缝1 mm, 接收狭缝0.16 mm, 步进式扫描, 步宽0.04° , 扫描速度为2° · min-1, 扫描范围为5° ~70° 2θ 。
高温原位XRD测试采用Holland Panalytical X'Pert-Pro MPD仪器, 将脱矿煤粉(CM130N和BC210)放在高温原位样品台器件上、 N2氛围下检测, 每升高一个温度或者保持一段时间, 实时进行XRD扫描检测, Cu靶, 管压36 kV, 管流30 mA, 步进式扫描, 扫描速度5° · min-1, 扫描范围为5° ~70° 2θ 。 分别从25、 200、 500、 650、 800、 1 000和1 200 ℃保温10 min测得原位XRD谱图。
1.2.2 高分辨透射电镜(HRTEM)
脱矿煤粉与乙醇混合稀释后滴到铜网, 待乙醇挥发完毕后, 使用Jeol JEM 2100F 场发射透射电子显微镜, 进行形貌与002衍射测试。
1.2.3 X射线光电子能谱(XPS)
XPS测定利用Thermo ESCALAB 250XI型X射线光电子能谱仪完成。 X射线源: 电压是16 kV, 电流是14.9 mA, 束斑直径是650 μ m; 通能(pass energy): 全谱是100 eV, 步长0.5 eV; 窄谱元素高分辨谱是30 eV, 步长0.05 eV。
1.2.4 拉曼光谱(Raman)
采用LabRam HR Evolution型拉曼光谱仪, Ar+激光激发, 激光波长514 nm, 在各脱矿煤粉的3~6个不同位置进行测试, 再将所测结果取平均值。
1.2.5 傅里叶红外光谱(FTIR)
采用溴化钾压片法, 称取煤粉(未脱矿处理)和溴化钾粉末按照质量比1∶ 200比例混合, 在玛瑙研钵中充分研磨混匀, 置于模具中, 在油压机上压成透明薄片, 烘箱中100 ℃持续干燥4 h。 采用美国生产的Thermo Scientific Nicolet iS5型FTIR光谱仪, 将干燥后薄片固定后置于样品室中, 设置波数测定范围4 000~400 cm-1, 累加扫描次数为32次, 分辨率为4 cm-1。
1.2.6 紫外-可见光-近红外漫反射测试(UV-Vis-NIR)
采用日本Shimadzu UV-3600i Plus光谱仪, 在200~2 500 nm 范围内进行光吸收测量。
2.1.1 XRD结构特征
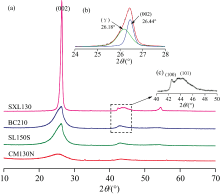
在煤系石墨的XRD谱中[图1(a)], ~26° 2θ 、 ~42° 2θ 、 ~44° 2θ 、 ~54° 2θ 附近分别存在对应石墨晶体(002)、 (100)、 (101)、 (004)晶面的特征峰[5]。 通常认为XRD谱图中~26° 2θ 处的宽缓峰是由(002)峰和γ 带叠加产生[图1(b)], 前者归属于类石墨的微晶结构(或缩聚芳香核), 后者为无定形碳或微晶边缘的脂肪族侧链[6]。 随着变质程度升高, ~44° 2θ 处的叠合峰(10l)分裂为(100)峰和(101)峰[图1(c)], (100)峰变高变窄, 芳香层片尺寸增大, 宽缓的(101)峰是煤系石墨微晶层间配置开始ABAB…规则排列的结果。 (10l)重叠峰的叠合和过度展宽可能是由微晶的大小和形状、 旋转、 微应变和缺陷结构(如堆垛层错)等因素引起[2]。 通过拟合分峰求解子峰峰位、 半峰宽等数据(表2), 并由Scherrer公式和Bragg方程求取芳香层面网间距(d002)、 堆砌度(Lc)和延展度(La)等结构参数, 由Willliamson-Hall方法计算微晶颗粒平均尺寸(crystallite size, Dsize)以及微观应变(microstrain, ε strain)[7, 8]。 面网间距(d002)在0.347 2~0.336 8 nm 之间, 石墨化度DG在0~0.837之间。 按照煤系石墨鉴定标准[1], CM130N属于高变质无烟煤, SL150S和BC210属于半石墨, SXL130属于煤系石墨。
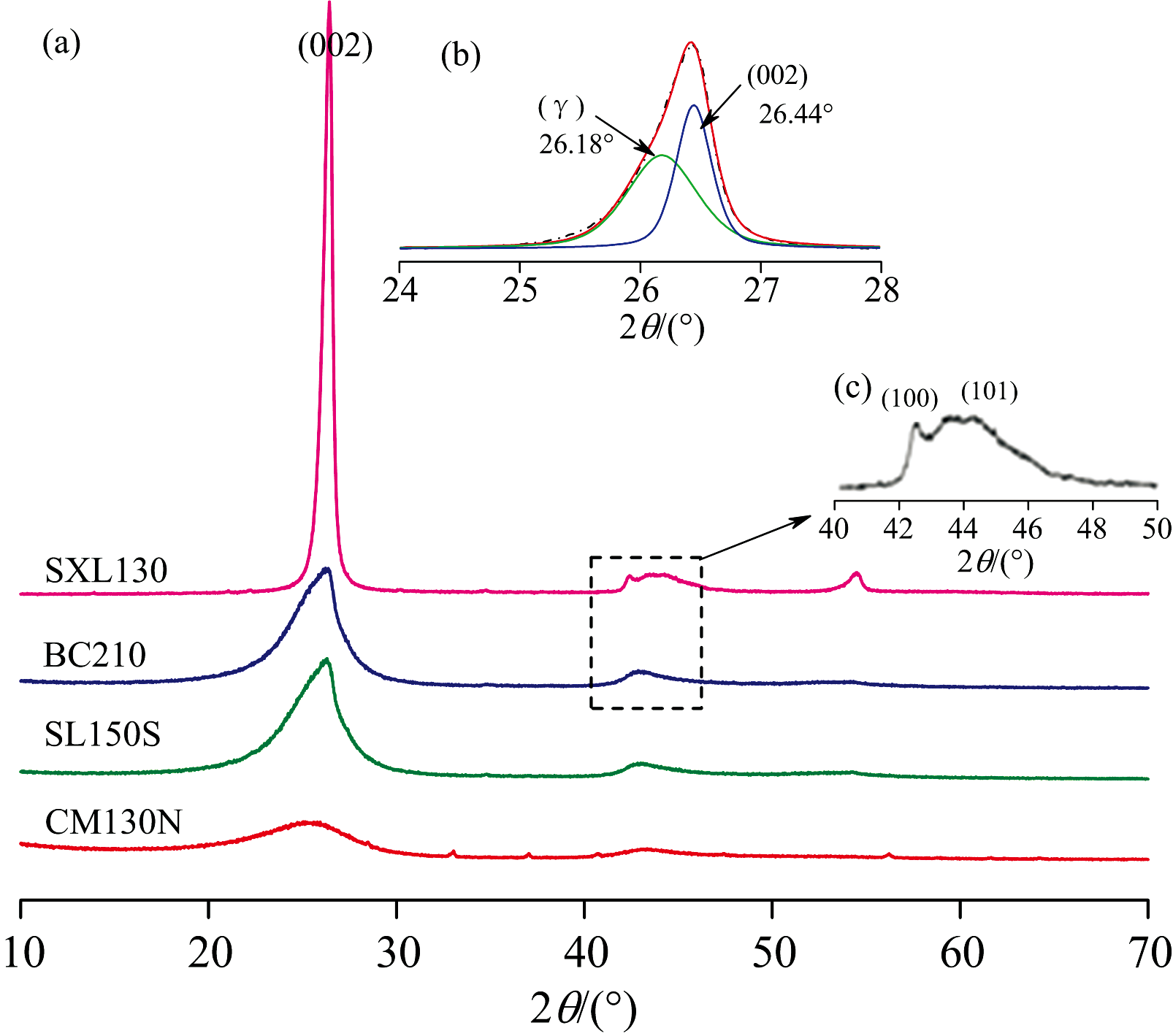 | 图1 (a) 煤系石墨的XRD谱图; (b) (002)衍射峰分峰拟合; (c) (100)和(101)峰分裂Fig.1 (a) XRD patterns of coal measures graphites; (b) Curve-fitting of (002) band; (c) (100) and (101) peaks were moderately separated |
| 表2 (γ )、 (002)峰的峰位和半峰宽以及XRD结构参数 Table 2 Peak positions, FWHM, and obtained structural parameters extracted from curve-fitting of the (002) and (γ ) bands |
石墨化前期的晶格构建准备阶段(CM130N), (002)衍射峰较宽缓, 含有无序碳形成的紊层结构, 微晶尺寸较小(表2), 取向随机[2]; 随着石墨化作用增强, 煤系石墨芳香层片的堆砌高度(Lc)增大(Lc> 10 nm), 有序畴增大, 仍呈紊层堆积, 为雏晶形成阶段; 在高温主导, 剪应力作用参与下, 煤系石墨微晶尺寸以及沿c轴方向堆砌高度的明显增大, d002减小趋于完善有序石墨结构的面网间距, 石墨微晶旋转、 择优取向, 微观应变(ε strain)减小(表2), 衍射峰逐渐变窄, 且数值趋于理想石墨的值。 因此, 煤系石墨热膨胀行为亦会表现出更明显的各向异性, 即c轴方向的热膨胀主导。 石墨化程度的提高减少了结构缺陷, 使得热膨胀系数更加稳定和可预测。
2.1.2 HRTEM特征
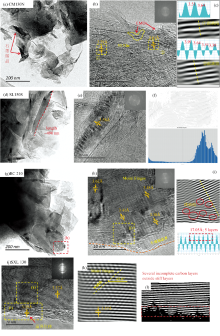
利用高分辨透射电镜可研究煤系石墨微晶结构的尺寸、 芳香层面网间距及有序度。 煤系石墨的演化阶段可划分为芳层石墨(芳层结构)、 微柱石墨(微柱结构)、 柔绉石墨(柔绉结构)与平直石墨等四个阶段[9]。
高变质无烟煤(CM130)芳香层片呈鳞片状[图2(a)], 具有局部定向域(LMOs), 可见数层堆叠, 延伸长度不稳定[图2(b)], 芳香层片间距3.58~3.61 Å [图2(c)], 主要结构缺陷类型有孔洞缺陷、 堆叠缺陷、 碳层的不规则弯曲以及碳层取向位错缺陷等。 散乱分布的芳层结构与石墨微晶结构是完全不同的, 是石墨化过程的准备阶段(开端), 称为芳层石墨。 选区电子衍射图上, 衍射环呈月牙状图[图2(b)], 表明芳香层片有择优取向。 XRD谱图(002)衍射峰呈鼓包, d002=0.347 2 nm。
 | 图2 高变质无烟煤-煤系石墨纳米结构特征Fig.2 Nanostructural characteristics of meta-anthracite-coal measures graphite |
半石墨(SL150S、 BC210)中可观察到芳香层片的集合体呈鳞片状[图2(d, g)], 刚性的芳香层片物理旋转和再定向[图2(e, f)], 芳香层片由于旋转而产生小的相对错位, 重叠产生Moiré 条纹[图2(h)], 位相差达37.40° ; (002)衍射环明显, (10l)和(004)衍射环初现分离[图2(h)], 标志着煤中芳香层片的有序性和各异向性均有显著发展; 芳香层片迅速增大, 延展几十至数百纳米[图2(d, g)], 芳香层片堆叠大于20层, 层间距为3.38~3.62 Å [图2(d, h)], 逐渐接近理想的石墨结构, 但存在堆叠缺陷、 位错等结构缺陷[图2(i)], 与柔绉石墨阶段相当。 XRD谱图(002)衍射峰呈驼峰状, d002为0.339 3~0.338 8 nm, (100)和(101)衍射峰仍未完全分立。
煤系石墨(SXL130)中平直有序芳香层片[图2(j)]的(002)衍射环明锐, (10l)和(004)衍射环已经分开, 明亮对称的光斑显示石墨晶格发育, 弥散环为层内缺陷或边缘; 芳香层片延展至上百纳米, 芳香层片堆叠达数十层, 层间距为3.36~3.35 Å , 为相对完好的石墨晶体, 与平直石墨阶段相当。 但局部存在Moiré 条纹[图2(k)], 并且在石墨微晶表面或边缘还出现了部分以乱层结构形式存在的无定形碳[图2(l)], 且这两种结构在HRTEM衍射图所表现出来的衍射点阵有明显区别, 既有清晰规整的衍射点阵又有衍射强度相对减弱的衍射环。 换言之, 即使在相对完好石墨化煤样中, 未经解体的衍射环与结构缺陷依然存在, 这种不同衍射域的差异与不均匀的石墨化程度相关。
2.1.3 Raman特征
拉曼光谱作为一种优异的表面无损探测技术, 成为分析煤系石墨的有效手段[2]。 石墨的层状结构决定了石墨的振动模式, 即平面内振动, 碳原子在石墨层内的振动模式; 平面外振动, 碳原子垂直于石墨层的振动模式[5]。 在拉曼光谱图中(图3), 位于1 300~1 400 cm-1的峰, 称为D峰(Defect peak), 与石墨中的缺陷或无序结构有关[2]。 D峰可分解为多个子峰(D1, ~1 350 cm-1; D2, ~1 620 cm-1等), 反映不同缺陷类型和结构特征, 其中, D1峰源于各种无序结构的A1g振动模式, 表征短暂模式的sp2环。 位于1 500~1 600 cm-1的峰, 称为G峰(Graphitic peak), 其源于石墨结构中C— C的E2g伸缩振动模式, 表征环状和链状的sp2原子团[2]。 煤系石墨按照微晶结构有序度的增加可分为3个阶段: ①无定形碳(无烟煤)至高变质无烟煤阶段, 对应于从链状结构逐渐变为环状结构的sp2碳, 并且环的缩合数不断增大; 链状结构的键长比环状结构的键长短, 导致振动频率减小。 在拉曼光谱上表现为G峰位置从≈ 1 600 cm-1向低波数移动, ID1/IG最大至≈ 3。 ②半石墨阶段, 结构持续有序化, 各向异性显现, G峰位置向低波数移动, ID1/IG继续减小; ③煤系石墨阶段, G峰位置稳定在1 575 cm-1附近, ID1/IG显著降低, 接近理想石墨晶体结构时, ID1/IG< 0.5或更低。
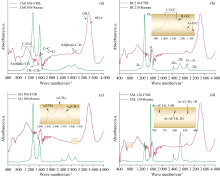
 | 图3 煤系石墨的傅里叶红外光谱和拉曼光谱Fig.3 Fourier infrared spectroscopy and Raman spectroscopy of coal measures graphite |
| 表3 煤系石墨Raman结构参数 Table 3 Raman spectroscopy parameters of coal measures graphite |
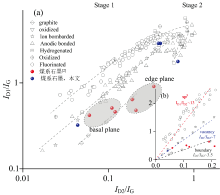
图3中拉曼光谱均显示明显的D1峰和D2峰以及2D1峰。 其中D2峰(G峰的高频肩峰)为边缘诱导的结构缺陷[2]。 各向异性煤系石墨的ID1/IG均小于0.5, AD1/AD1+D2+G均小于0.6[1, 2]。 无烟煤阶段, 边缘结构不明显, D2峰或许不可见。 半石墨阶段, 边缘结构开始形成且逐渐显著, D2峰的强度增加; 石墨阶段, D2峰强度达到最大值, 但随着石墨化程度的进一步提高, D2峰强度可能会略有下降, 这是因为石墨晶体尺寸增大, 边缘结构相对减少。 在低缺陷密度下, ID1/IG与ID2/IG参数之间的线性依赖关系[10], 并且不同类型缺陷对应的ID1/ID2比值不同(图4)。 因此可以借鉴ID1/ID2来识别煤系石墨结构缺陷的性质, 即与sp3杂化相关缺陷的ID1/ID2最大(≈ 13), 空位型缺陷的ID1/ID2减小(≈ 7), 石墨中边缘型缺陷的ID1/ID2最小(≈ 3.5)[10, 11]。 粉末样品中高缺陷密度(ID1/IG)的CM130N、 SL150S和BC210[图4(a), 蓝色点]与缺陷类型没有吻合(Stage 2), 以至于缺失了有关缺陷性质的任何信息, 仅SXL130呈现空位型缺陷。 各向异性煤系石墨[2]边缘与基面缺陷相比, 基面具有更高的ID1/ID2[图4(b), 红色点]。 这使得拉曼光谱成为全面表征石墨有序性的有力工具。
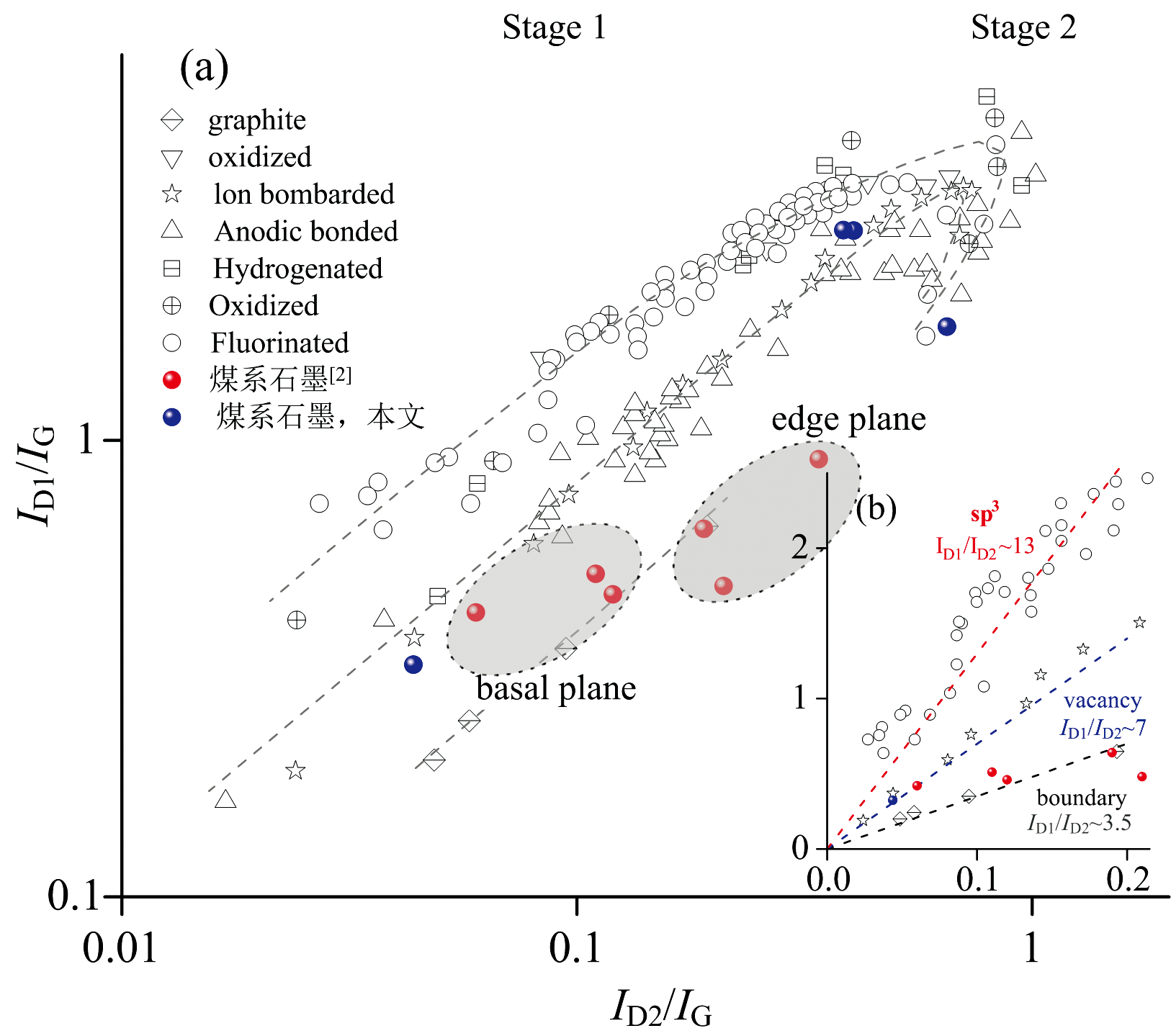 | 图4 用于判别缺陷类型的ID1/IG与ID2/IG比值的关系(据文献[10]修改)Fig.4 The relationship between ID1/IG and ID2/IG ratio for identifying defect types (According to reference [10]) |
2.1.4 FTIR特征
完美石墨晶体本身几乎不具有红外活性, 在FTIR光谱中通常没有明显的吸收峰[5]。 石墨的对称性高, E2g模是红外非活性的[12]。 图3(a)显示, 1 350 cm-1(D1峰)与石墨中的缺陷和无序结构相关, 对应在红外光谱中表现为1 000~1 800 和2 800~3 000 cm-1处较宽吸收峰, 显示石墨微晶基面或者边缘存在某些化学杂质。 1 580 cm-1(G峰)在红外光谱中不显著, 但在拉曼光谱中是石墨特征峰。
煤系石墨红外光谱中, 虽然煤系石墨的对称性高, 导致许多振动模式在红外光谱中不活跃, 但红外光谱仍然可提供关于石墨结构缺陷表面官能团的重要信息。 3 435 cm-1处强烈吸收峰表示自缔合的羟基形成的氢键, 在3 618 cm-1处的弱肩峰是羟基和π 键形成的氢键(图3)。 2 800~3 000 cm-1的脂肪结构吸收峰, 归因于脂肪烃CH、 — CH2和— CH3伸缩振动[图3(c)]。 1 700~1 770 cm-1处的弱吸收峰, 可能来自酮、 醛、 羰和羧基振动。 1 620 cm-1的吸收峰归因于氢键缔合的羰基, 具有— O— 取代的C═C。 1 580 cm-1处的吸收峰归因于石墨碳层中芳环伸展, 并随变质程度升高, 其吸收峰强度降低。 1 385和1 465 cm-1的吸收峰分别归因于CH3对称弯曲振动、 脂肪族CHx变形振动。 1 000~1 300 cm-1范围的吸收峰表示酚、 醇、 醚、 酯的C— O振动, 中心峰位为1 095 cm-1归因于C— O— C伸缩振动及Si— O伸缩振动[图3(a, b)]。 700~900 cm-1主要代表芳环中C— H取代变形振动, 苯环的取代方式为邻位二取代和邻位三取代[图3(d)]。 通过结合拉曼光谱和红外光谱的分析, 可以更全面理解煤系石墨的物理化学性质。
2.1.5 XPS特征
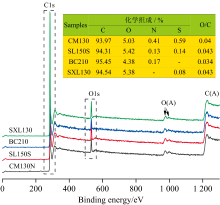
X射线光电子能谱(XPS)信息来自样品表面几个至几十个原子层, 可用来分析煤系石墨表面的元素组成和官能团含量[13, 14]。 为使样品更接近实际情况, 未对煤粉进行脱矿处理。 煤系石墨表面以C、 O为主要组成元素(图5), 还含有少量的N、 S元素。 随着石墨化程度增高, 其表面N、 S元素越低, 尤其是SXL130表面甚至检测不到N元素。
 | 图5 煤系石墨的XPS全谱Fig.5 XPS spectrum of coal measures graphite |
煤系石墨的C(1s)精细谱采用Gaussian-Lorentzian函数进行分峰拟合(图6), 六个子峰结合能的归属依据文献[13]归纳如下: 284.4 eV, Graphite C— C; 285.0 eV, Hydrocarbons C— H; 286.1~286.3 eV, Alcohol C— OH/ether O— C; 287.6~287.7 eV, Carbonyl C═O; 288.6~289.1 eV, Carboxyl COOH/ester COOR; 290.5~290.8 eV, Carbonate CO3/Satellite Π — Π * ), 进一步分析还可以得到煤系石墨表面的元素组成以及表面官能团的相对含量。 由图6 和表4可知, 石墨化度对表面C、 O含量及其结合形式影响不大。 多种含氧官能团同时存在, 且含量较低, 给谱峰解叠带来难度。 其中, C— C占含碳官能团的59.27%~68.54%, C— O占含碳官能团的8.07%~14.04%(表4)。 细小鳞片状石墨微晶颗粒之间及其基面内部的缺陷, 刚性表面的不完整炭层堆叠形成结构缺陷, 以及石墨微晶的边缘缺陷, 这些位点是含氧官能团主要集中区域。 相对而言, 边缘结构缺陷处的含氧官能团更为集中, 而基面内缺陷处的含氧官能团分布较为分散, 从而会影响石墨微晶各向的表面性质。
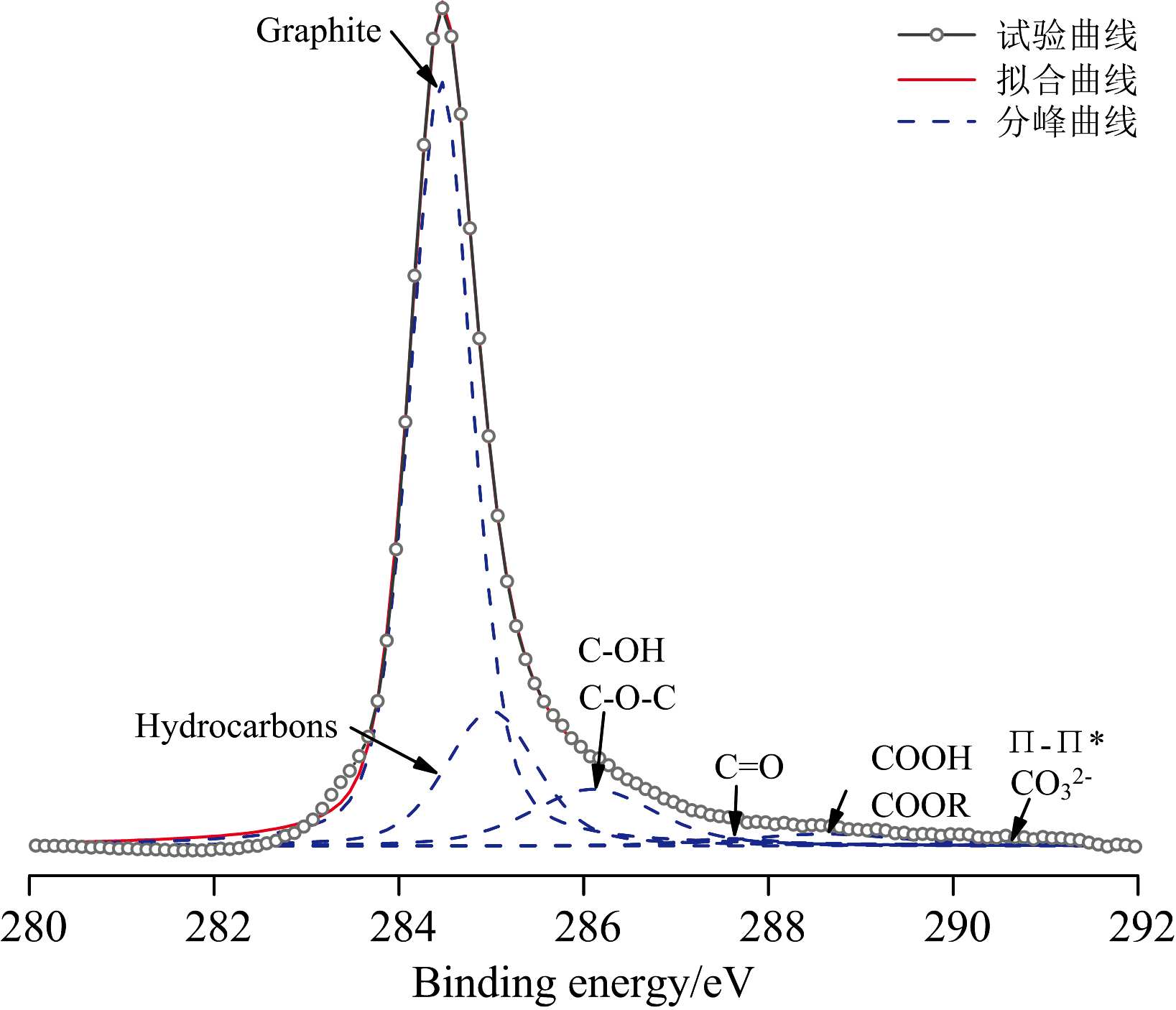 | 图6 煤系石墨XPS的C(1s)谱拟合峰Fig.6 XPS C(1s) spectrum fitting peak of coal measures graphite |
| 表4 煤系石墨的XPS C(1s)拟合结果 Table 4 XPS C(1s) fitting results of coal measures graphite |
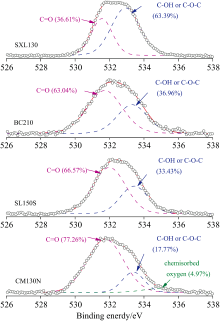
和C(1s)分析相一致, 煤系石墨的O(1s)精细谱[14]可分解为: 531.2~531.6 eV, C═O groups; 532.8~533.1 eV, C— OH and/or C— O— C— groups; 535.4~535.8 eV, chemisorbed oxygen and/or adsorbed water。 由图7 可知, 煤系石墨中C— O官能团含量高于C═O官能团, 这与C(1s)谱中表现相一致。 随着石墨化度增高, 煤系石墨中的氧元素含量降低(表1), 石墨微晶表面羰基C═O的比例逐渐减少, 而C— OH and/or C— O— C— 相对增多(图7)。 这可能由于C═O官能团的较高反应活性, 在石墨化过程中更容易被还原或脱除而含量降低, 相对于C— O官能团较快。
 | 图7 煤系石墨XPS的O(1s)谱拟合峰Fig.7 XPS O(1s) spectrum fitting peak of coal measures graphite |
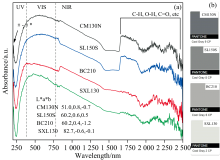
实验测得表1中4种煤样在波长200~2 500 nm范围的紫外-可见光-近红外吸收光谱曲线(图8), 所有光谱曲线形状相似, 但石墨化程度高的样品在近红外区域变得更加平坦。 在波长190~360 nm范围的紫外区, 石墨烯强吸收峰在270 nm附近[15], 其表示芳香烃离域π 电子的跃迁, 苯环的π — π * 跃迁通常在254 nm附近, 而煤系石墨中仍存在缺陷或杂质, 可能会使吸收峰向短波长移动(蓝移)至208 nm附近[图8(a)]。 在煤系石墨化过程中, 缩聚芳香核排列由无序向有序的转变, 表现为离域电子数增多, 导致π — π * 跃迁所需能量降低, 吸收强度增加。
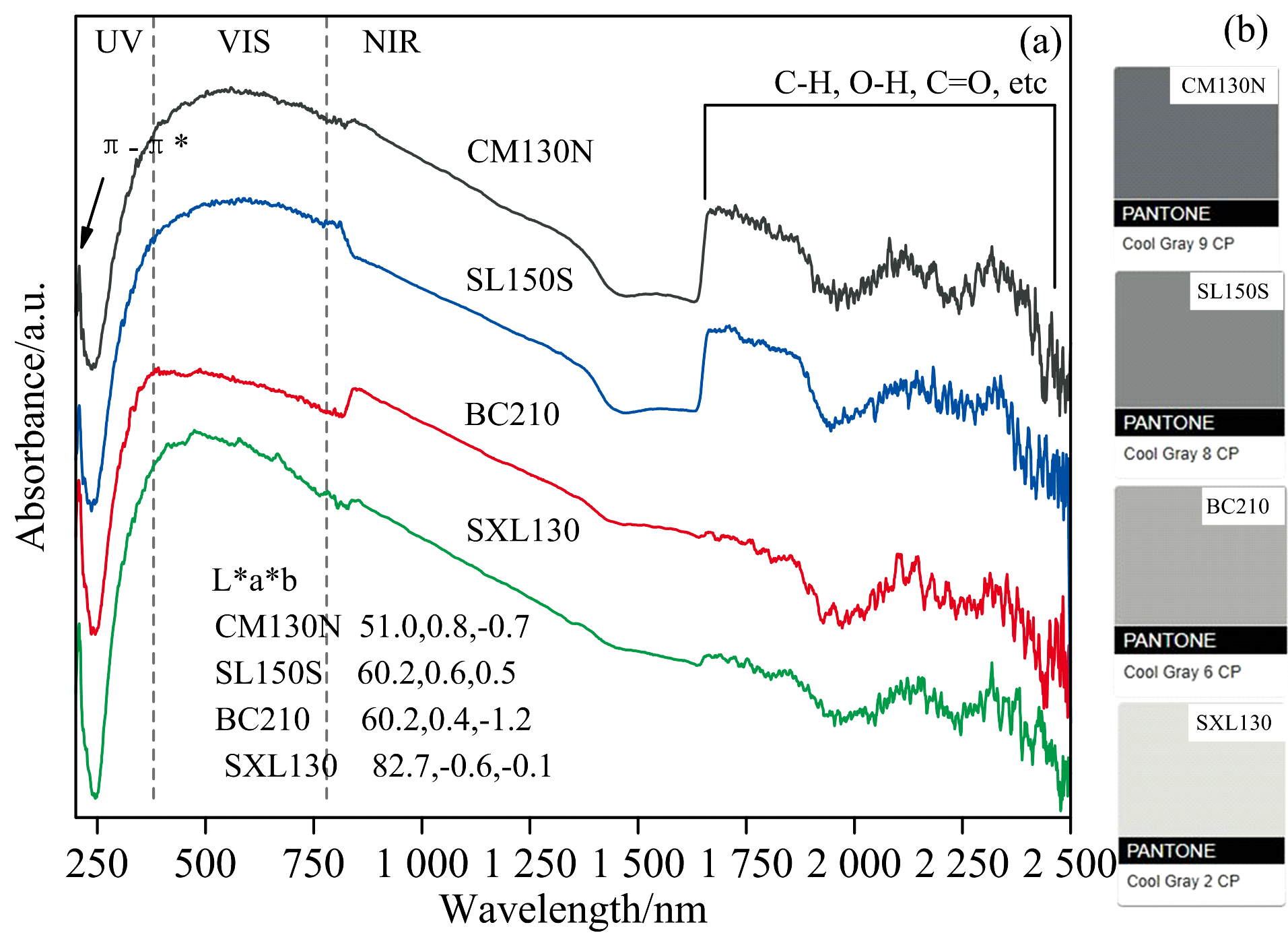 | 图8 煤系石墨的紫外-可见光-近红外吸收光谱Fig.8 Ultraviolet-Visible-near infrared absorption spectra of coal measures graphite |
波长380~780 nm范围为可见光区, 主要与分子或原子的电子能级跃迁相关。 煤系石墨颜色变化是有机组分表面和内部各类光线的综合作用的反映, 石墨化作用阶段, 涡层状叠片结构逐渐向石墨微晶结构过渡, 微晶尺寸和反射率急剧增加, 各向异性显著。 利用Colortell在线颜色工具(https://www.colortell.com/colortool), 将380~780 nm光谱数据转化为CIE1976 L* a* b* 色彩空间数值, 对获取的L* 、 a* 、 b* 值进行颜色仿真[图8(b)], 选择了色差Δ E76(ab)最小值的仿真颜色— — 潘通PANTONE Cool Gray。 4种煤样仿真颜色分布于Cool Gray 9 CP至2CP; 亮度(L* )体现颜色的强弱(单位投影面积上的反光强度), 随着石墨化作用的增加, 其本身物理、 化学结构变化导致相关反光能力增加, 如SXL130的L* 最高达82.7。
波长780~2 500 nm范围的近红外区代表分子振动基频的倍频和合频的谱带, 主要涉及C— H、 C═O和O— H的伸缩振动的合频或倍频[16]。 其中, 1 400和1 900 nm处的吸收峰与自由水、 结合水或吸附水有关; 2 100~2 600 nm范围的吸收峰可归因于黏土矿物的金属— OH倍频以及C— H伸缩振动的合频和倍频。 此外, 在1 700和2 300~2 500 nm范围有时也会出现较弱的条带, 这些条带对应于各种脂肪族侧链C— H伸缩振动、 含氧官能团的倍频和合频。 石墨化程度越高, 表面结构越完善, 780~2 500 nm吸收峰越不明显[图8(a)]。 这可能是因为更完善的表面结构减少了分子振动的无序性, 从而降低了吸收峰的强度。
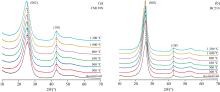
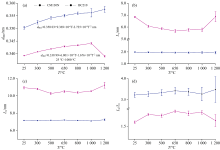
图9为CM130N(石墨化度DG=0)和BC210(石墨化度DG=0.605)在不同温度下(从室温到1 200 ℃)的XRD谱图。 不同温度处理下得到的XRD图谱非常相似, 均显示出典型的非晶炭材料的衍射峰特征。 其中, (002)衍射峰反映了微晶结构中芳香层片在c轴方向的堆叠情况, 峰越窄越高, 表明芳香层片堆叠越规整; (10l)衍射峰则反映了芳香层片的尺寸大小, 峰越窄越高, 说明芳香层片越大。
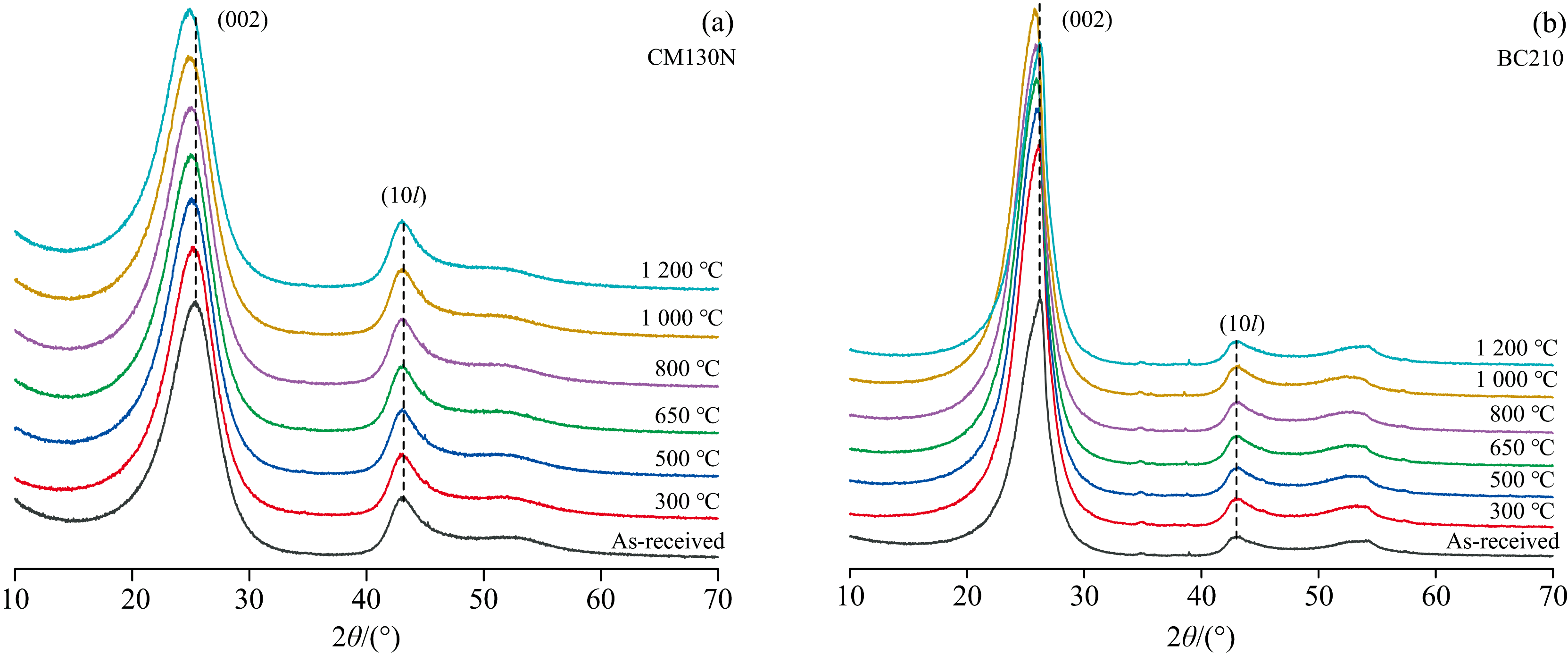 | 图9 不同温度处理的煤系石墨002反射峰向低角度方向移动Fig.9 Shifts to lower angle side of 002 reflections for coal measures graphite treated at various temperatures |
随热处理温度升高, CM130N的(002)峰位从原煤的25.41° 2θ 逐步减小至1 200 ℃的24.88° 2θ [图10(a), 表5], 表明芳香层片间距d002逐渐增大, 结构趋于松散, 这可能是高温下煤中的脂肪侧链和含氧官能团逐渐分解, 释放的小分子气体导致层间距增大。 与此同时, 微晶堆垛高度Lc、 芳香层片直径La变化不显著[图10(b, c)], 这可能是因为其初始结构较为规整, 热处理过程中未发生显著的重排。
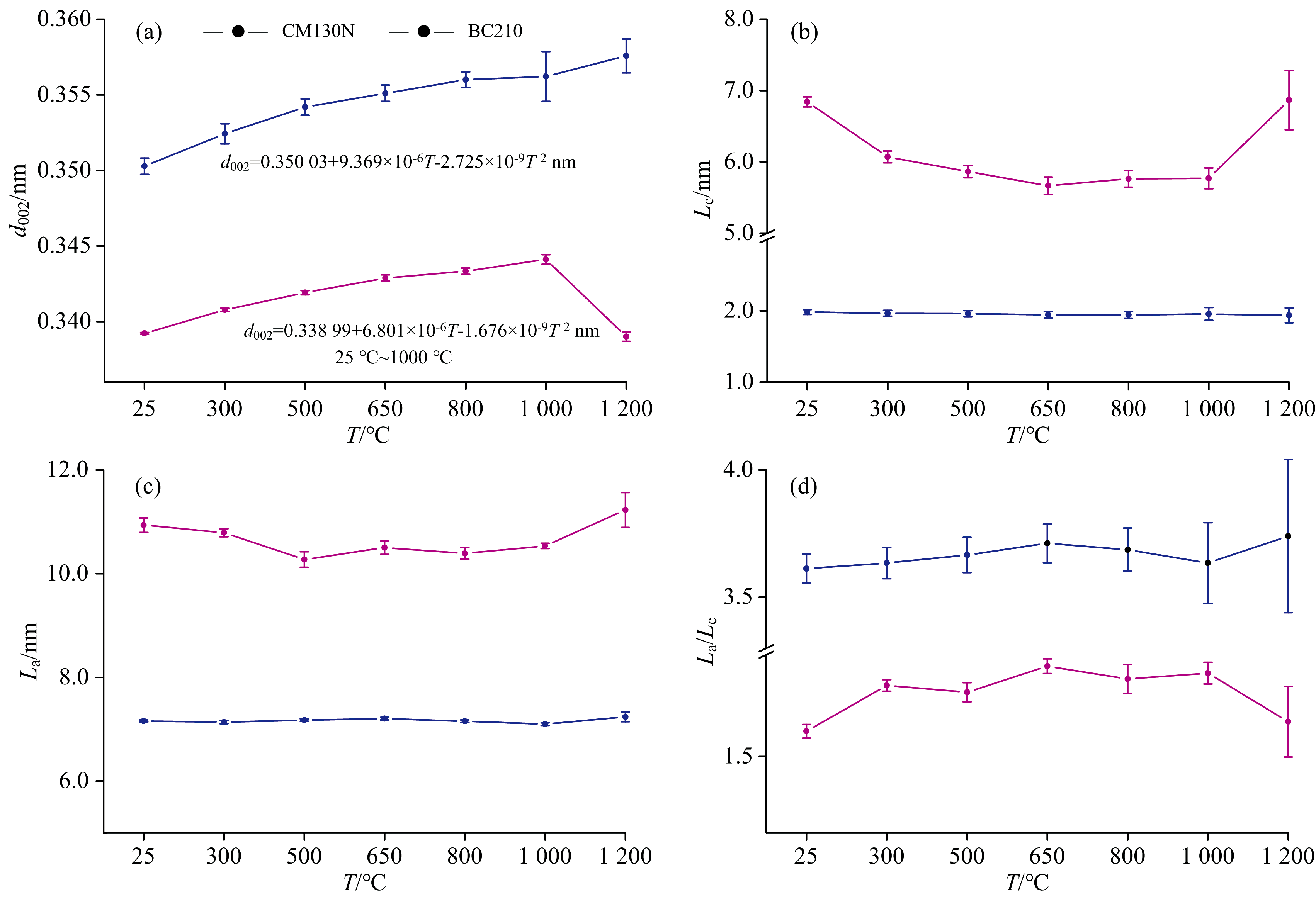 | 图10 不同温度XRD结构参数d002、 La、 Lc和La/Lc的演化趋势Fig.10 Evolution trend of XRD structural parameters d002, La, Lc and La/Lc at various temperatures |
| 表5 煤系石墨的高温原位XRD结构参数 Table 5 High temperature in-situ XRD structural parameters of coal measures graphite |
相比之下, BC210的(002)峰位从原煤的26.25° 2θ 逐步减小至1 000 ℃的25.87° 2θ , 但在1 200 ℃时又增大至26.27° 2θ [图10(b), 表5]。 其芳香层片间距d002呈现出先增大后减小的趋势[表5, 图10(a)], 这种变化可能与样品的初始结构有关, 热处理过程中发生重排。 在高温下, 缩聚反应使芳香层片堆叠更加紧密。 此外, Lc和La从原煤分别减小到500 ℃时的5.86和10.27 nm, 500 ℃后几乎保持不变[图10(b, c)]。 这可能是由于样品中的脂肪侧链和含氧官能团大量分解, 导致微晶结构的堆叠高度和尺寸减小。 直到1 000 ℃ 后, 随着热处理过程的深入, 芳香层片通过缩聚反应重新排列, 在1 200℃时, Lc和La分别达到6.86和11.23 nm。 样品的初始结构和化学组成显著影响其热处理过程中的结构演变路径。
石墨的热膨胀系数具有明显的各向异性, 平行于基平面方向的热膨胀系数比垂直于基平面方向的要小[4]。 随着热处理温度的升高, 石墨微晶在弱结合c方向上的膨胀(表5)是由层间距的等效膨胀来调节的, 而在a方向上, 随温度升高形成晶格缺陷的迹象, 从而降低了平均微晶尺寸(W-Hsize)。 尽管在所考虑的温度范围内, La、 Lc、 W-Hsize和ε strain随温度的变化不大, 但石墨化程度高的BC210随温度均呈现先降低后增高的变化规律。 ε strain来源于石墨化未完成而残留的“ 乱层” 扭曲, 以及各向异性热膨胀在微晶内部及晶界处产生的“ 热致应变” 。 随着热处理增大, 出现600~700 ℃ 的“ ε strain最小值平台” (表5), 之后ε strain的重新上扬可能由“ 非结构本征” (空位、 层错等缺陷)导致。 整体上, 热处理使d002值增大, 微观应变ε strain释放而协同减小。
石墨微晶具有显著的各向异性, 层间方向的结合力弱, 而在层内方向的共价键结合力较强, 其力学性质在不同方向上表现出差异, 晶体沿不同取向的弹性模量不同; 而在石墨成长过程中, 由于冷却速率不均匀或生长速率差异, 可能会在微晶内部产生微观应力, 不同取向的应变能不同。 然而, 石墨微晶的应变产生部位与生长模式(柱面生长、 分支生长等)密切相关, 如柱面生长, 应变主要集中在晶体的轴向或径向(图11箭头处), 而分支生长, 应变更容易集中在分支的连接部位或尖端。 因此, 可推断石墨化的驱动力是微晶边界处产生的微观应力, 这些微观应力导致芳香层片的褶皱, 这些微观应力通过热处理(或变质程度升高)被消除时, 即会诱导石墨微晶重新排列、 甚至生长。
 | 图11 煤系石墨微晶边缘处纳米结构特征Fig.11 Nanostructure characteristics at the edge of graphite crystallites in coal measures graphite |
石墨颗粒之间的相互作用对微观应变有着重要影响。 应力传递、 晶界和位错的作用、 微晶尺寸和形状等因素可能均会影响微观应变的大小和分布。 自然演化序列无烟煤-半石墨-煤系石墨微观应变规律性变化。 高变质无烟煤CM130N的结构中主要由芳香族碳骨架组成, 含有少量的脂肪侧链和含氧官能团(图3), 内部微观应变相对较小。 石墨化初期, 碳原子开始形成层状结构, 层间距较大, 层间排列不够规则, 微晶尺寸较小, 内部应力集中点较多, 微观应变增加(表2)。 随着石墨化程度增高, 石墨微晶结构逐渐优化, 碳原子排列更加有序, 内部应力逐渐释放, 微观应变开始减小。
各向异性煤系石墨的热膨胀系数在不同方向上表现出显著差异。 层内碳原子间的键长会随着温度升高(300~1 200 ℃)而增加, 导致层内方向的膨胀, 但高变质无烟煤CM130N的La变化不明显, 可能源于脂肪侧链和含氧官能团的键断裂, 阻碍微晶径向生长, 但半石墨BC210的La呈先减小后增大。 随着温度的升高, XRD谱图中(002)衍射峰向高角度方向移动, 显示层间距离增加, 而XRD谱图的半峰宽变化不明显, 显示微观应变相对较小, 但BC210的Lc表现为先减小后增大, 可能在温度作用下, 微晶重新排列, 沿着c轴方向生长。
(1)XRD和Raman结果表明, 煤系石墨微晶尺寸、 沿c轴方向堆砌高度随着石墨化作用增强而显著增大, 各向异性特征显现。 不同类型缺陷对应的ID1/ID2比值不同, 与各向异性煤系石墨边缘面缺陷相比, 基面具有更高的ID1/ID2, 煤系石墨结构缺陷的性质以空位型缺陷、 边缘型缺陷为主。
(2)HRTEM结果显示高变质无烟煤芳香层片具有局部定向域, 可见数层堆叠, 延伸长度不稳定, 在煤系石墨微晶表面或边缘还出现了部分以乱层结构形式存在的无定形碳, 选区衍射图既有清晰规整的衍射点阵又有衍射强度相对减弱的衍射环。
(3)XPS和FTIR结果显示, 细小鳞片状石墨微晶颗粒之间及其基面内部的缺陷, 刚性表面的不完整炭层堆叠形成结构缺陷, 以及石墨微晶的边缘缺陷, 这些位点是含氧官能团主要集中区域。 随着石墨化度增高, 煤系石墨中的氧元素含量降低, 石墨微晶表面羰基C═O的比例逐渐减少。 1 580 cm-1(G峰)在红外光谱中不显著, 却是石墨的拉曼光谱特征峰。
(4)UV-Vis-NIR光学特征显示, 因煤系石墨中存在多环芳烃, π — π * 跃迁吸收峰略微蓝移至208 nm附近。 4种煤样仿真颜色分布于Cool Gray 9 CP~2 CP, 随着石墨化作用的增加, 其本身物理、 化学结构变化导致反光能力增加。
(5)石墨的热膨胀系数具有明显的各向异性, 平行于基平面方向的热膨胀系数比垂直于基平面方向的要小。 随着温度的升高, 石墨微晶在弱结合c方向上的膨胀是由层间距的等效膨胀来调节的, 而在a方向上, 随温度升高形成晶格缺陷的迹象, 从而降低了平均微晶尺寸(W-Hsize)。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|