
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
钴铱基电催化剂酸性析氧反应的原位表面增强拉曼光谱研究
[胡策军2  , 胡艳芳
, 胡艳芳1, * , 谢微3, * ]
, 胡艳芳, 谢微]
|
|


作者简介: 胡策军, 1995年生,福州大学材料科学与工程学院副教授 e-mail: cejun_hu@fzu.edu.cn
酸性析氧反应(OER)是质子交换膜电解槽的核心过程, 但其强腐蚀性环境与复杂的反应路径使高效催化剂的开发面临严峻挑战。 揭示真实反应条件下催化活性位点的动态演化规律与反应机理, 对设计高稳定性酸性OER催化剂具有重大科学意义。 采用置换法制备了高活性、 高稳定性的CoIrO x酸性OER催化剂, 并以此为模型组装成兼具拉曼光谱增强和催化性能的双功能结构, 实现了钴铱基催化剂在酸性OER中表界面微环境的原位监测。 电动力学测试结果表明, 在催化剂活化过程中, 催化剂表面钴原子不断浸出产生空位, 更多铱位点被暴露并被氧化为Ir5+活性相, 从而促进反应。 结合原位SERS和H/D同位素对照实验确认了在酸性体系下该反应的中间物种为OOH。 采用18O标记法进一步探索催化表面微结构的动态变化, 发现CoIrO x催化剂的Ir—O键振动峰在OER电位下出现了明显的蓝移, 成功证实CoIrO x催化剂的OER过程遵循晶格氧氧化机制(LOM)。 这项工作对钴铱基催化剂的反应机制提出了新的见解, 为OER反应路径的探索开辟了思路。
The acidic oxygen evolution reaction (OER) is a core process in proton exchange membrane water electrolyzers. However, the highly corrosive environment and complex reaction pathways pose significant challenges for developing efficient catalysts. Understanding the dynamic evolution of catalytically active sites and the reaction mechanism under realistic operating conditions is crucial for designing highly stable acidic OER catalysts. In this work, highly active and stable CoIrO x catalysts were prepared via a displacement method. Electrokinetic studies revealed that during catalyst activation, cobalt leaching generates surface vacancies, which in turn expose iridium sites that are oxidized to the active Ir5+ phase, thereby promoting the reaction. In situ surface-enhanced Raman spectroscopy (SERS) combined with H/D isotope experiments confirmed that OOH is the key intermediate species in the acidic OER process. Furthermore,18O isotope labeling was used to probe the dynamic changes in the catalyst's surface microstructure. A pronounced blue shift in the Ir—O vibrational peak at OER potentials indicated that the OER on CoIrO x follows a lattice oxygen oxidation mechanism (LOM). This work provides new insights into the reaction mechanisms of CoIr-based catalysts and offers a novel approach for exploring OER pathways.
酸性电解体系与碱性体系相比具有欧姆损失低, 高电流密度大, 电压效率高, 气体纯度高, 系统设计紧凑, 系统响应快等诸多优点[1, 2], 工业化生产中主要是基于质子交换膜(PEM)电解槽的酸性体系电解水。 作为水分解的半反应, 析氧反应(OER)涉及了缓慢的四电子转移动力学过程, 在很大程度上决定了能量转移效率。 研究者们已经开发了许多具有超高活性和稳定性的非贵金属催化剂用于碱性析氧反应, 但在恶劣的酸性环境中, 这些过渡金属基氧化物/氢氧化物很容易发生溶解或表面结构转变, 导致催化性能急剧下降[3, 4]。 目前, 只有IrO2和RuO2可以作为酸性OER的基准催化剂[5], 而价格昂贵, 资源短缺等问题限制了它们的大规模应用。
掺杂非贵金属组分已被证明是减少Ir, Ru等贵金属含量, 降低成本的有效途径。 在酸性条件下, 贵金属虽然占比相对较少, 但仍起到催化OER的主要作用。 掺杂的组分可以与贵金属发生电子相互作用, 引起催化剂内电荷分布、 晶格参数和轨道状态的变化[6, 7]。 其中一种掺杂策略是向催化剂中添加一些耐酸的惰性材料[8](如W、 Sn、 Nb和Ti等)来减少贵金属的含量并保持整体催化剂稳定性, 然而, 此类材料通常表现出与纯Ir氧化物相似的性能; 另一种是掺杂Fe, Co, Ni, Cu, Cr等在酸性环境中不稳定的金属元素, 借助它们的浸出效应来改善催化剂活性[9]。 在最近的一项研究中, Bajdich, Jaramillo等发现掺杂不同元素表现出不同的活性, 顺序如下: IrTi< IrSn2< Ir< IrNi< IrCr, 其中IrCr具有最突出的性能。 这是由于Cr在电化学过程中浸出后提供了一个富Ir的表面, 并且掺杂位点上有比Ir位点更优的氧结合能, 因此掺杂后获得了增强的催化活性[9]。
此外, 为设计合成高效且稳定的酸性催化剂, 需要在催化机制方面进行深入研究。 虽然研究者们对碱性OER的相关机理的认识已经取得了较大的进步, 但受到酸性催化剂短缺的限制, 对酸性OER的反应机理却知之甚少。 目前, 公认的两种OER催化机理有两种, 分别是吸附物种演变机制(AEM)和晶格氧氧化机制(LOM)[10]。 AEM机制中, 在酸性和碱性两种条件下机理看似不同, 但都包含四个基本的步骤和相同的活性中间体(OH* , O* , OOH* ), Rossmeisl等发现Δ G* OOH与Δ G* OH之间的线性关系, 导致热力学理论上的过电势为370 mV, 从而限制了催化剂性能的进一步提高[11]。 与传统的AEM机理不同, LOM机理通过引入晶格氧的方式创造相邻的两个位点, 打破了中间物种吸附能之间的线性关系。 在LOM途径中, 催化剂表面随着氧的析出过程发生动态变化, 因而处于热力学不稳定状态。 因此, 了解催化剂的工作路径对于催化机理的阐述以及高效催化剂的设计具有十分重要的意义。
基于此, 将少量的贵金属铱元素掺杂到CoOx中形成CoIrOx来构筑高活性和高稳定性的催化剂, 并以此作为模型监测了钴铱基催化剂的动态演变过程。 结合原位表面增强拉曼光谱研究以及同位素替代实验探究了酸性OER中的活性中间体组成, 并使用18O标记法证实该催化剂经历了LOM机制, 为酸性催化剂的OER路径探究提供了一种新的研究范式。
钴铱氧化物(CoIrOx): 基于氧化钴纳米颗粒的合成方法, 首先将128 mg的乙酰丙酮钴(Ⅱ )放入在三颈烧瓶中, 升温至50 ℃并在剧烈搅拌下将其溶解于10 mL的油胺, 在N2气氛下将混合物加热至120 ℃并保持50 min, 然后快速升温至250 ℃并保持1 h以初步裂解生成CoOx纳米颗粒。 与此同时, 在另一三颈烧瓶中, 将50 mg Ir(acac)3溶解在3 mL 油胺(OAm)中, 在120 ℃同样保持50 min后升温至250 ℃, 待升至温度, 将Ir— OAm复合物迅速注入至CoOx的合成体系中, 继续在250 ℃熟化40 min, 通过热裂解产生的Ir与Co的置换作用, 形成最终的CoIrOx纳米颗粒。 待产物冷却至室温后, 用正己烷和乙醇洗涤3次, 得到的纳米颗粒重悬在正己烷中备用。
80 nm金纳米颗粒(Au NPs)的合成[12]: 首先将150 mL, 2.2 mmol· L-1的柠檬酸钠溶液放置在250 mL三颈烧瓶中升温至120 ℃, 待溶液沸腾后, 向其中注入1 mL 25 mmol· L-1的氯金酸溶液, 溶液的颜色由黄色变为蓝灰色最后变为淡粉色, 即得到粒径为10 nm左右的金种子。 随后开始生长步骤, 将体系温度降至90 ℃, 加入1 mL 25 mmol· L-1的氯金酸溶液保持30 min后, 再次加入1 mL 25 mmol· L-1的氯金酸溶液继续保持30 min。 至此完成一次生长步骤, 取出55 mL溶液, 加入2 mL 60 mmol· L-1柠檬酸钠和53 mL 去离子水, 使总体积保持不变。 将此溶液继续作为种子, 重复整个过程, 直至得到80 nm的金纳米颗粒。
金二氧化硅核壳结构(Au@SiO2)的合成[13]: 取1 mL 80 nm Au NPs, 离心(270 g, 15 min)弃上清液后重悬于1 mL H2O中。 依次加入10 μ L, 1%的柠檬酸钠溶液和10 μ L 10%(3-巯基丙基)三甲氧基硅烷(MPTMS)的乙醇溶液。 超声混匀后, 放置在50 ℃, 750 rpm的温度控制混匀仪上孵育1 h, 然后按照之前的转速离心, 重悬于1 mL水中。 最后, 在悬浮液中加入7 μ L, 0.054%的硅酸钠溶液, 再放置在温控混匀仪上, 以90 ℃, 750 rpm保持1 h, 得到的Au@SiO2胶体经离心再分散于1 mL异丙醇中备用。
双功能超级结构(Au@SiO2@CoIrOx)的组装[14]: 采用范德华力组装方法, 将合成的Au@SiO2 NPs分散在异丙醇中, CoIrOx作为卫星NPs分散在正己烷中。 然后, 在超声下将1 mL的CoIrOx NPs注入到1 mL的异丙醇Au@SiO2胶体中。 10 min后, 混合胶体颜色由砖红色变为蓝色表示超级结构组装成功。 随后将超级结构离心(200 g, 10 min), 弃上清液后, 分散在乙醇中备用。
电化学测试: 首先进行浆液的配制, 将离心清洗好的1 mg 催化剂分散在1 mL N, N-二甲基甲酰胺(DMF)中, 加入1 mg导电炭黑, 经超声分散均匀后, 加入10 μ L Nafion粘合剂, 超声1 h后即可得到用于电化学测试的浆液。 测试时采用三电极体系, 将测试浆液滴加到玻碳电极表面, 烘干后作为工作电极, Ag/AgCl电极和铂丝分别作为参比电极和对电极, pH 1的H2SO4水溶液为电解液。 在收集电化学数据之前, 首先以10 mV· s-1的扫描速率在相对于可逆氢电极(RHE)0.9~1.6 V之间循环多次以获得稳定的性能, 随后在5 mV· s-1的扫描速率下采集循环伏安曲线(CV)或线性扫描伏安曲线(LSV)。 实验过程中未对溶液电阻引起的IR降进行补偿。 电化学阻抗谱(EIS)测量是通过施加5 mV的交流电压, 频率范围为105~10-1 Hz, 外加相对于RHE 1~1.7 V的偏压进行的, 在测试完成后通过Zview软件进行等效电路图的拟合, 拟合结果在图2(f)中展示。
表面增强拉曼光谱(SERS)测试: 将组装的双功能纳米颗粒制备成单层膜结构, 将原位池的玻碳电极倒置于界面处, 单层膜结构自然吸附在电极表面, 晾干后组装原位池。 将pH 1的H2SO4作为电解液充入原位池中, 排出气泡后连接电化学工作站即可进行电化学原位测试。 在同位素替代实验中, 分别使用重水和H
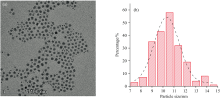
CoIrOx纳米颗粒通过热裂解法合成, 借助CoOx种子将铱的金属离子还原, 同时还原出来的Ir与Co之间发生置换反应, 最大化地利用Ir前驱体, 得到掺杂含量较高的CoIrOx纳米颗粒。 如图1所示, 得到的纳米颗粒形貌均匀, 尺寸均一, 约为10.5 nm, 最终纳米颗粒中Co和Ir的比例约为3:1。
 | 图1 (a)CoIrOx纳米颗粒的透射电镜图(标尺: 100 nm); (b)50个CoIrOx纳米颗粒的粒径分布柱状图Fig.1 (a) HRTEM image of CoIrOx NPs (Scale bar: 100 nm); (b) Histogram of the particle size distribution of 50 CoIrOx NPs |
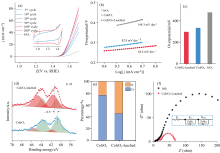
随后, 我们测试了CoIrOx催化剂在pH 1的硫酸中的析氧反应活性。 从图2(a)可以看出, CoIrOx性能和稳定性都远超于商业化的IrO2, 并且随着循环次数的增加, 催化剂的性能越来越好。 除此之外, 位于1.05/1.0和1.35/1.4 V两处分别对应于Ir3+/Ir4+和Ir4+/Ir5+的氧化还原峰也逐渐明显, 且非法拉第电容区变宽, 这表示随着测试时间的延长, 催化剂的电化学比表面积(ECSA)逐渐增大, 循环后的ECSA接近于之前的1.5倍, 我们猜测这可能由于Co在酸性环境下浸出, 催化剂表面有更多的Ir位点暴露, 导致最终性能的提升。 从图2(b)和(c)中可以看出Co浸出后的催化剂拥有三者中最小的Tafel斜率和过电位(10 mA· cm-2电流密度下), 表明浸出后的CoIrOx催化剂的动力学最快, 性能最好。
 | 图2 (a)不同循环次数下采集的CoIrOx的线性扫描伏安曲线, 灰色曲线为商业化IrO2的电化学性能, 插图中使用第3圈和第200圈的CV曲线表示两对氧化还原峰的变化; (b)商业化IrO2, 以及浸出前后CoIrOx三种催化剂的Tafel斜率; (c)三种催化剂在10 mA· cm-2电流密度下的过电位; (d)电化学循环前后CoIrOx的XPS谱图; (e)电化学循环前后的CoIrOx催化剂中Co与Ir的元素占比; (f)浸出后CoIrOx和IrO2在pH 1的硫酸体系中的Nyquist图Fig.2 (a) Linear sweep voltammetry (LSV) curves of CoIrOx collected at different cycle numbers; The inset shows the variation of two pairs of redox peaks using the CV curves of the 3rd and 200th cycles; Tafel slopes (b) and overpotentials (c) of commercial IrO2 and CoIrOx catalysts before and after leaching; (d) XPS spectra of CoIrOx before and after electrochemical cycling; (e) Elemental composition in CoIrOx catalysts before and after electrochemical cycling; (f) Nyquist plots of leached CoIrOx and IrO2 in a pH 1 sulfuric acid system |
为确认测试过程中Co浸出这一猜想, 将CoIrOx进行100圈的循环测试之后, 采用ICP测试催化剂中Co, Ir两元素含量。 结果发现, 在循环过后催化剂中的Co与Ir的比例由原来的3:1转变为接近1:1, 此外, XPS结果也表现出同样的现象, 在循环之后位于780 eV左右的Co(2p)峰强度降低, 而在60 eV左右的Ir(4f)峰的相对强度增大, 经峰面积计算可得出Co/Ir的相对比例变化与ICP测试结果一致[图2(d)]。 上述实验结果表明, 在酸性OER的过程中CoIrOx中的Co确实会从氧化物中浸出, 从而暴露出富Ir的表面进行催化。
为进一步获得CoIrOx在OER过程中的电子转移和反应动力学, 我们使用原位电化学阻抗(EIS)进行测试。 基于EIS数据拟合出了最佳的等效电路图, 如图2(f)所示, 其中, Rs为溶液电阻, CPE1为双层电容, Rct与界面电荷转移反应有关, CPE2和Rp与电极内膜的介电性能和电阻有关。 在CoIrOx中, 当电压高于1.5 V时, Rct的值迅速降低, 这与LSV测试得到的OER起始电位一致, 说明在这个电压下发生了OER氧化反应, 而IrO2则在更滞后的电位1.6 V时才出现这一现象, 表明相对于IrO2来说, CoIrOx催化的OER反应速率更快。 接下来我们使用电化学-原位SERS光谱来探究CoIrOx催化剂高活性的本质原因。
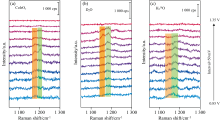
原位SERS光谱测试基底采用范德华力组装方法制备, 将CoIrOx纳米颗粒分散在正己烷中, 与分散在异丙醇中的Au@SiO2纳米颗粒混合, 超声组装成Au@SiO2@CoIrOx核-卫星超级结构, 该超级结构的透射电镜表征如图3(a)所示。 随后, 将超级结构制备成单层膜转移至玻碳电极表面作为工作电极, 以pH 1的H2SO4为电解液, Ag/AgCl和Pt丝分别作为参比电极和对电极进行原位电化学-拉曼光谱测试。
 | 图3 (a)Au@SiO2@CoIrOx超级结构的元素分布图, 右上角为对应的单颗粒透射电镜图(标尺: 20 nm); (b)Au@SiO2@CoIrOx超级结构表面采集的原位SERS谱图Fig.3 (a) Elemental mapping images of Au@SiO2@CoIrOx; The inset shows TEM image with a scale bar of 20 nm; (b) In situ surface-enhanced Raman spectroscopy (SERS) spectra acquired on the Au@SiO2@CoIrOx superstucture surface |
由电化学性能测试结果可知, CoIrOx的电催化OER过程可以分成三个阶段。 施加电压之后, 0.9~1.2 V范围内, 大多数的Ir3+转变为Ir4+。 随后在1.35 V被继续氧化为Ir5+活性中间体, 最终催化OER过程的发生。 在对应的原位SERS光谱中, 也可以观察到同样的趋势。 如图3(b)所示, 在开路电压下, 可以观察到三个比较明显的峰, 位于620 cm-1 的δ 峰归属于Ir3+的Ir— O键的伸缩振动, 而位于510和750 cm-1的γ 和ε 两个峰归属于四价Ir4+的Ir— O键的伸缩振动[15]。 随着电压的升高, 这δ 和ε 两个峰在1.25 V逐渐消失并伴随着β 、 θ 和η 三个峰的出现, 这表示Ir位点的氧化进入了下一阶段。 根据文献报道, β 和θ 两个峰分别对应于Ir5+的Ir— O键的弯曲振动和伸缩振动, 而对于η 峰的归属, Pavlovic等[16]通过DFT计算的方式, 将位于829 cm-1处的η 峰归属于活性中间体Ir=O的伸缩振动, 同时Alexander J Cowan等[17]使用氘代的电解液进行原位测试, 该峰位置并没有发生明显的移动, 说明这一振动模式不太可能是质子化的氢氧化物或过氧化物, 因此将η 峰归属于高价Ir5+的Ir— O的伸缩振动。 除上面所述的这几个峰之外, 在电压升到1.45 V时, 580 cm-1处出现了一个较强的振动峰, 图中标记为“ * ” , 这与文献中金红石IrO2在563 cm-1处的峰相近, 可能由于催化剂在OER过程中生成了不同相的氧化铱。 最后, 我们将上述峰位置和峰的归属总结在表1中。
| 表1 OER条件下CoIrOx表面SERS谱图中各峰归属 Table 1 Vibrational peaks in the SERS spectra acquired on the CoIrOx surface under electrochemical OER conditions |
除上述催化剂的变化, 在SERS谱图的1 100~1 300 cm-1范围内, 还捕捉到了在1 178和1 203 cm-1处有中间体的信号, 但位置相对碱性中的(1 150和1 167 cm-1)向高波数偏移, 为确认中间物种成分, 我们进行了同位素替代实验。
氘代和18O代的实验与原位电化学测试过程相同, 只是将电解液替换为重水或H
 | 图4 以pH 1硫酸的(a)H2O, (b)D2O和(c)H218O溶液为电解液的Au@SiO2@CoIrOx超级结构表面采集的原位SERS谱图Fig.4 In situ SERS spectra acquired on the Au@SiO2@CoIrOx using: (a) H2O, (b) D2O, and (c) H218O solutions (pH 1) as electrolytes |
同位素的拉曼频率与正常实验中测到的拉曼频率之比可以通过质量公式计算得到
$\begin{aligned} \gamma & =\frac{v(\mathrm{O}-\mathrm{OD})}{v(\mathrm{O}-\mathrm{OH})}=\frac{\frac{\sqrt{m(\mathrm{O})+m(\mathrm{OD})}}{\sqrt{m(\mathrm{O}) \times m(\mathrm{OD})}}}{\frac{\sqrt{m(\mathrm{O})+m(\mathrm{OH})}}{\sqrt{m(\mathrm{O}) \times m(\mathrm{OH})}}} \\ & =\frac{\frac{\sqrt{16+18}}{\sqrt{16 \times 18}}}{\frac{\sqrt{16+17}}{\sqrt{16 \times 17}}}=98.64 \% \end{aligned}$(1)
经过计算, 若为OOH物种, 则在氘代后的理论拉曼频率为1 162和1 186 cm-1, 与我们测得的位置极为接近, 也就说明该物种为OOH。 与此同时, 我们还使用H218O做了进一步验证, 在图4(c)中我们发现中间物种同样向低波数移动, 同样的, 经过质量公式进行计算
$\begin{aligned} \gamma & =\frac{v\left(\mathrm{O}^{18}-\mathrm{O}^{18} \mathrm{H}\right)}{v\left(\mathrm{O}^{16}-\mathrm{O}^{16} \mathrm{H}\right)}=\frac{\frac{\sqrt{m\left(\mathrm{O}^{18}\right)+m\left(\mathrm{O}^{18} \mathrm{H}\right)}}{\sqrt{m\left(\mathrm{O}^{18}\right) \times m\left(\mathrm{O}^{18} \mathrm{H}\right)}}}{\frac{\sqrt{m\left(\mathrm{O}^{16}\right)+m\left(\mathrm{O}^{16} \mathrm{H}\right)}}{\sqrt{m\left(\mathrm{O}^{16}\right) \times m\left(\mathrm{O}^{16} \mathrm{H}\right)}}} \\ & =\frac{\frac{\sqrt{18+19}}{\sqrt{18 \times 19}}}{\frac{\sqrt{16+17}}{\sqrt{16 \times 17}}}=94.43 \% \end{aligned}$(2)
因此, 若为OOH物种, 则18O替代后的理论拉曼频率为1 112和1 136 cm-1, 然而在谱图中却没有发现在这两个位置左右的振动峰, 反而在相对于计算结果稍高一点波数的1 148 和1 176 cm-1两处观测到两个形状类似的峰, 这表明中间物种中包含氧原子但没有全部被电解液中的18O替代, 因此我们猜测OOH中间物种中只有一个来自于水, 而另一个可能来源于催化剂的晶格氧。 若按上述猜测, 只计算一个18O,
$\begin{aligned} \gamma & =\frac{v\left(\mathrm{O}-\mathrm{O}^{18} \mathrm{H}\right)}{v\left(\mathrm{O}-\mathrm{O}^{16} \mathrm{H}\right)}=\frac{\frac{\sqrt{m(\mathrm{O})+m\left(\mathrm{O}^{18} \mathrm{H}\right)}}{\sqrt{m(\mathrm{O}) \times m\left(\mathrm{O}^{18} \mathrm{H}\right)}}}{\frac{\sqrt{m(\mathrm{O})+m\left(\mathrm{O}^{16} \mathrm{H}\right)}}{\sqrt{m(\mathrm{O}) \times m\left(\mathrm{O}^{16} \mathrm{H}\right)}}} \\ & =\frac{\frac{\sqrt{16+19}}{\sqrt{16 \times 19}}}{\frac{\sqrt{16+17}}{\sqrt{16 \times 17}}}=97.41 \% \end{aligned}$(3)
这一计算结果对应的波数为1 147和1 172 cm-1, 与我们测到的位置接近, 因此我们将对催化剂的晶格氧氧化机制进行深入探索。
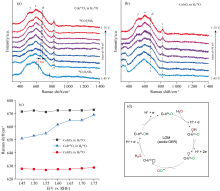
为验证OER过程中催化剂的晶格氧是否与电解液中的O进行了交换, 我们依然采用同位素标记的方法。 首先, 将CoIrOx催化剂放置在pH 1的硫酸H218O溶液中, 施加足够高的电压(1.75 V)使催化剂中的16O全部被标记为18O, 从而得到高价的CoIr18Ox, 随后将电解液替换成pH 1的硫酸
 | 图5 标记后的CoIr18Ox(a)和CoIrOx标记前(b)在pH 1的硫酸H216O溶液中采集的电化学原位SERS谱图; (c)不同样品在不同电解液中CoIrOx的θ 拉曼峰位置; (d)推测的CoIrOx催化OER机理示意图Fig.5 In situ SERS spectra of isotopically labeled CoIr18Ox (a) and CoIrOx (b) in pH 1 sulfuric acid-based H216O solution; (c) Raman peak position of the θ for CoIrOx across different samples and electrolytes; (d) Proposed reaction mechanism of CoIrOx-catalyzed OER based on experimental evidence |
对于每个电位下的偏移程度, 在1.75 V时观察到最显著的位移, 与H216O中的峰位置只相差几个波数, 其中Ir— O 的θ 峰蓝移了37 cm-1, 约为完整同位素交换41 cm-1完全位移的90%[图5(c)], 造成同位素替代不完全的原因可能有以下几点: (1)在催化剂上并不是所有的位点都是有活性的; (2)电压升高以后催化剂中更多的Co浸出从而暴露出更多的Ir位点; (3)我们在每个电压下电解时间为10 min, 可能时间过短而不足以完成全部替代。 根据同位素标记实验, 我们得出结论: CoIrOx催化的OER过程中确实有晶格氧的参与。
基于上述实验, 将推测的CoIrOx催化剂在OER过程中的反应机理总结在图5(d)中。 与传统的Ir基催化剂经历的AEM机制不同, 我们所合成的CoIrOx催化剂经历LOM机制。 首先, 在酸性条件下, 随着电压升高, 水分子经过脱质子过程转变为OH吸附在催化剂表面, 随后经过进一步氧化并与催化剂中的晶格氧形成氧气脱离催化剂表面, 留下一个氧空位和还原态的Ir位点, 最终这些空位被水中的O补齐, 以进入下一次循环。
采用置换法制备了在酸性条件下相对稳定且具有高活性的OER催化剂-CoIrOx。 催化剂中Co不断浸出产生晶格空位并暴露出富Ir表面, 使催化剂性能随循环次数增加而逐渐提升。 随后, 借助SERS对整个电化学OER过程进行原位检测, 捕捉到了酸性OER过程中的活性中间体, 并采用D/18O同位素替代法, 明确该物种为OOH。 进一步地, 采用晶格氧置换方法探究了催化剂的反应路径, 得出OER过程中该催化剂遵循晶格氧氧化机制, 产物氧气中的两个氧原子一个来自水而另一个来源于催化剂中的晶格氧。 这一工作为OER过程的原子层面机制研究提供新思路。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|