

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
溶剂效应对2,2'-联吡啶-6,6'-二甲酸激发态分子内质子转移过程的影响
[古丽米热·亚尔麦麦提1, 2  , 宋鑫甜
, 宋鑫甜1 , 安桓1 , 布玛丽亚·阿布力米提1, * , 向梅1, * ]
, 宋鑫甜, 安桓, 向梅]
|
|


作者简介: 古丽米热·亚尔麦麦提,女, 1998年生,新疆交通职业技术学院机电工程学院专任教师 e-mail: 838811804@qq.com;宋鑫甜,女, 1999年生,新疆师范大学物理与电子工程学院硕士研究生 e-mail: 2979709323@qq.com
古丽米热·亚尔麦麦提,宋鑫甜:并列第一作者
采用含时密度泛函理论(TD-DFT)在Cam-b3lyp/6-31G(d、 p)理论水平上计算了2,2'-联吡啶-6,6'-二羧酸(BP6DC)在环己烷、 二氯甲烷和二甲基亚砜溶剂中的键长、 键角、 红外(IR)振动光谱、 最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)。 此外, 利用Multiwfn和VMD软件模拟了BP6DC在这3种不同溶剂环境中的空穴-电子轨道。 在实验层面, 通过稳态光谱仪测量了其吸收和发射光谱。 研究结果表明, 在环己烷(CYH)溶剂中, 由于BP6DC分子固有的对称性, 与两个氢键O12—H18…N10和O24—H25…N19相关的参数(键长、 键角)的变化是一致的。 相反, 在二氯甲烷(DCM)和二甲基亚砜(dimethyl sulfoxide, DMSO)溶剂中, 键长和键角的变化表现出相反的趋势。 通过势能面的分析, 溶剂极性对BP6DC分子激发态下氢键的影响效果得到了解释。 得出结论, BP6DC能够在环己烷溶液中发生激发态双质子转移。 相反, 在DCM和DMSO溶液中, 分子对称性被破坏, 导致只能发生单质子转移, 其单质子转移具有双通道特性。
Gulimire Yaermaimaiti and SONG Xin-tian: joint first authors
In this study, we used time-dependent density functional theory (TD-DFT) to calculate 2,2'-bipyridine-6 at the Cam-b3lyp / 6-31G (d, p) theoretical level. The bond length, bond angle, infrared (IR) vibration spectrum, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) of 2,2'-bipyridine-6,6'-dicarboxylic acid (BP6DC) in cyclohexane, dichloromethane, and dimethyl sulfoxide solvents were studied. In addition, the hole-electron orbitals of BP6DC in these three different solvent environments were simulated by Multiwfn and VMD software. At the experimental level, we measured its absorption and emission spectra using a steady-state spectrometer. Our results show that in cyclohexane (CYH) solvent, due to the inherent symmetry of the BP6DC molecule, the changes of parameters (bond length, bond angle) related to two hydrogen bonds O12—H18…N10 and O24—H25…N19 are consistent. On the contrary, in dichloromethane (DCM) and dimethyl sulfoxide (DMSO) solvents, the change of bond length and bond angle showed the opposite trend. Through the analysis of the potential energy surface, the effect of solvent polarity on the hydrogen bond in the excited state of the BP6DC molecule was explained. We conclude that BP6DC can undergo excited-state double proton transfer in cyclohexane solution. On the contrary, in DCM and DMSO solutions, the molecular symmetry is destroyed, resulting in only a single proton transfer, and this single proton transfer has dual channel characteristics.
氢键作为自然界最常见的弱相互作用之一, 是维持生命循环系统不可缺少的通道之一[1, 2]。 氢键在光化学, 光物理和光生物过程中也起着至关重要的作用[3, 4, 5]。 激发态质子转移作为生物和化学反应中最典型的酸碱中和反应, 主要是通过特定位点的氢键弱相互作用导致的反应[6, 7]。 氢键通常以X— H…Y的形式存在, 其中H原子带正电, 其余X, Y原子带负电, 这说明X— H是氢原子供体, H…Y是氢原子受体[8, 9]。 近年来研究者更青睐于对氢键的强度和与氢键相关质子运动的基础科学的研究[10, 11, 12]。 因此激发态质子转移过程成为近年的研究热点[13, 14, 15]。 20世纪中期, Taylor等首次报道了7-氮环及其双氢键二聚体的吸收与发射光谱, 并发现了激发态分子内双质子转移(excited state intramolecular biproton transfer, ESIDPT)[16], 这是一种非常典型的现象。 ESIDPT与激发态分子内质子转移(excited state intramolecular proton transfer, ESIPT)的区别是, 它通常由两个质子受体和两个质子供体组成, 即两个分子内氢键。 目前有大量实验研究ESIDPT过程, 观察到与ESIPT过程相似的双荧光峰[17, 18, 19, 20]。
2, 2'-联吡啶为含氮芳香杂合物, 其中两个N上有与稀土离子配位的能力[21]。 其广泛应用于荧光材料、 太阳能电池、 有机发光二极管[22, 23, 24]。 2, 2'-联吡啶的各种化合物受研究者的青睐, 其中具有两个分子内氢键的2, 2'-联吡啶-3, 3'-二醇被合成以来颇受关注[25, 26]。 最初1992年Pawel Borowicz等通过电光发射测量证明了其分子内双质子转移的结果[27]。 之后1996年Andrzej L Sobolewski等通过理论解释其双质子转移过程, 并且得到结论即通过计算势能面发现1Bu(Π Π * )发生无障碍的ESIPT[28]。 2021年Mohsen Oftadeh等研究了其取代基效应对ESIPT过程的影响[25]。 基于前人的结果我们重点研究2, 2'-联吡啶-6, 6'-二甲酸在激发态下的质子转移情况。 2, 2'-联吡啶-6, 6'-二甲酸用途广泛, 是生产液晶显示器和光电功能材料的重要材料。 2, 2'-联吡啶-6, 6'-二羧酸(2, 2'-bipyridine-6, 6'-dicarboxylic acid, BP6DC)与上述联吡啶分子的结构密切相关, 该分子同样有两个芳香吡啶环组成, 原则上可以形成平面反式或非平面顺式构象[29, 30]。 而质子转移主要是在溶液中进行的, 因此不得不考虑外界溶剂对BP6DC分子质子转移过程的影响。 因此本工作从溶剂效应这一角度全面研究了BP6DC分子的质子转移过程。 并且测量了BP6DC的吸收光谱和发射光谱, 利用DFT和TD-DFT 理论模拟计算了BP6DC分子的结构、 红外(infrared, IR)振动光谱、 空穴-电子轨道、 弱相互作用和势能等, 用以解释BP6DC分子ESIDPT过程。
紫外-可见吸收光谱用U-2450紫外-可见分光光度计(日本)测量。 FLS920稳态/瞬态荧光光谱仪(英国爱丁堡)测量荧光光谱。 该荧光光谱采用450 W的氦灯作为激发光源, 波长范围是200~900 nm。 实验中以230、 240和250 nm激发了分别溶解在环己烷(cyclohexane, CYH)环己烷, 二氯甲烷(dichloromethane, DCM)和二甲基亚砜(dimethyl sulfoxide, DMSO)中的BP6DC。 实验用试剂有BP6DC(纯度> 98%, 上海阿拉丁生化科技有限公司), 二甲基亚砜(无水溶剂, 上海阿拉丁生化科技有限公司), 环己烷, 二氯甲烷(纯度> 99.9%, 上海麦克林生化科技股份有限公司)。 使用前未进一步纯化。
所有理论计算都利用高斯16[31]程序完成。 运用不同的方法和基组进行模拟, 为了模拟吸收光谱的实验数据, 我们采用了可极化连续体模型(IEEPCM)[32, 33], 分别计算了BP6DC在CYH, DCM和DMSO三种溶剂中的吸收波长, 如表1所示。 对比发现, 基组Cam-b3lyp/6-31G(d、 p)的吸收波长计算结果与实验测得的吸收波长最吻合, 并且在结构优化过程中无虚频, 以上进一步证实结构的稳定性。 由于计算BP6DC分子时涉及氢键等长程相互作用, 因此添加了长程修正(coulomb-attenuating method)方法的基组的计算结果更接近实验值。 由于该分子比较简单, 因此只需加上对碳、 氮、 氢原子修正的d、 p轨道即可, 此计算结果也更接近实验值, 无需再添加额外的扩展基函数(“ +” )和更精细的高斯基函数(“ 311” )。 另外用Cam-b3lyp/6-31G(d、 p)基组计算分析了BP6DC在三种溶剂中的过渡态, 振动光谱(IR), 最高占据分子轨道(highest occupied molecular orbital, HOMOHOMO)和最低未占据分子轨道(lowest unoccupied molecular orbital, LUMO)和势能面。 此外为了更加透彻的分析BP6DC的激发态分子内质子转移过程我们运用Multiwfn程序[34]和VMD程序计算分子内弱相互作用, 空穴-电子轨道, 降低密度梯度(RDG)[35]。 此外, 我们还进行了BP6DC分子的红外光谱的实验, 然而我们所研究的是激发态下BP6DC分子的质子转移情况, 用到的计算方法为TD-DFT方法, 计算的是BP6DC分子激发态下的振动情况, 而我们实验所得到的红外光谱为基态下的振动情况, 所以两者差别较大。 由于不同的基组和方法对BP6DC分子的原子轨道进行了不同的描述, 导致BP6DC的化学键的力常数和折合质量不同, 从而影响了BP6DC分子的振动频率。
| 表1 利用TD-DFT方法结合不同基组计算了BP6DC在CYH, DCM和DMSO中的吸收波长 Table 1 The absorption wavelengths of BP6DC in CYH, DCM and DMSO calculated by TD-DFT method combined with different basis sets |
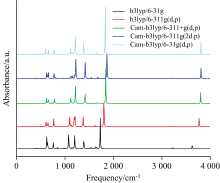
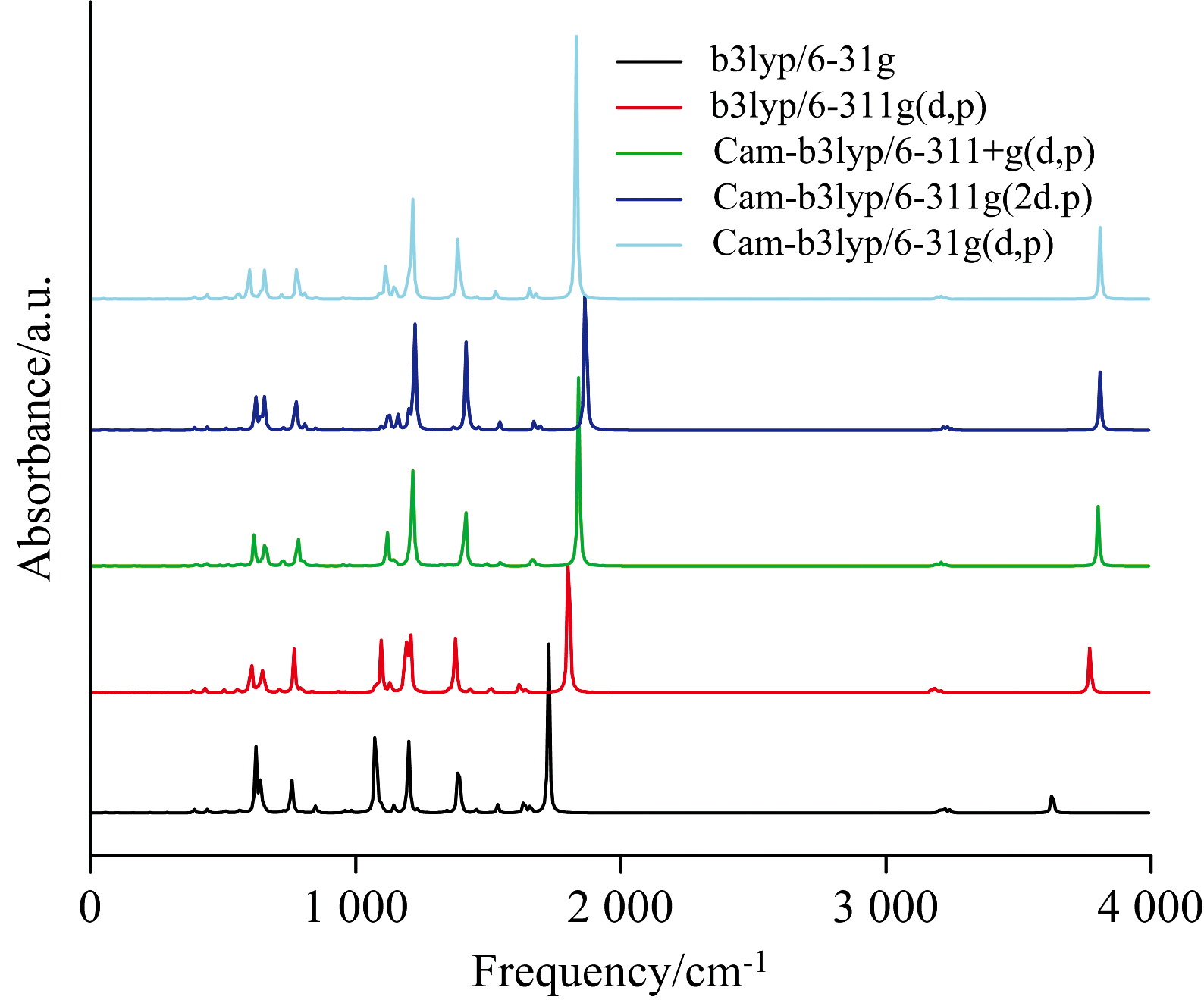 | 图1 利用TD-DFT方法结合不同基组计算了BP6DC的红外光谱Fig.1 The infrared spectra of BP6DC calculated by TD-DFT method combined with different basis sets |
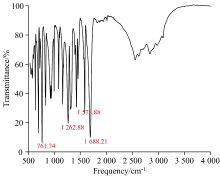
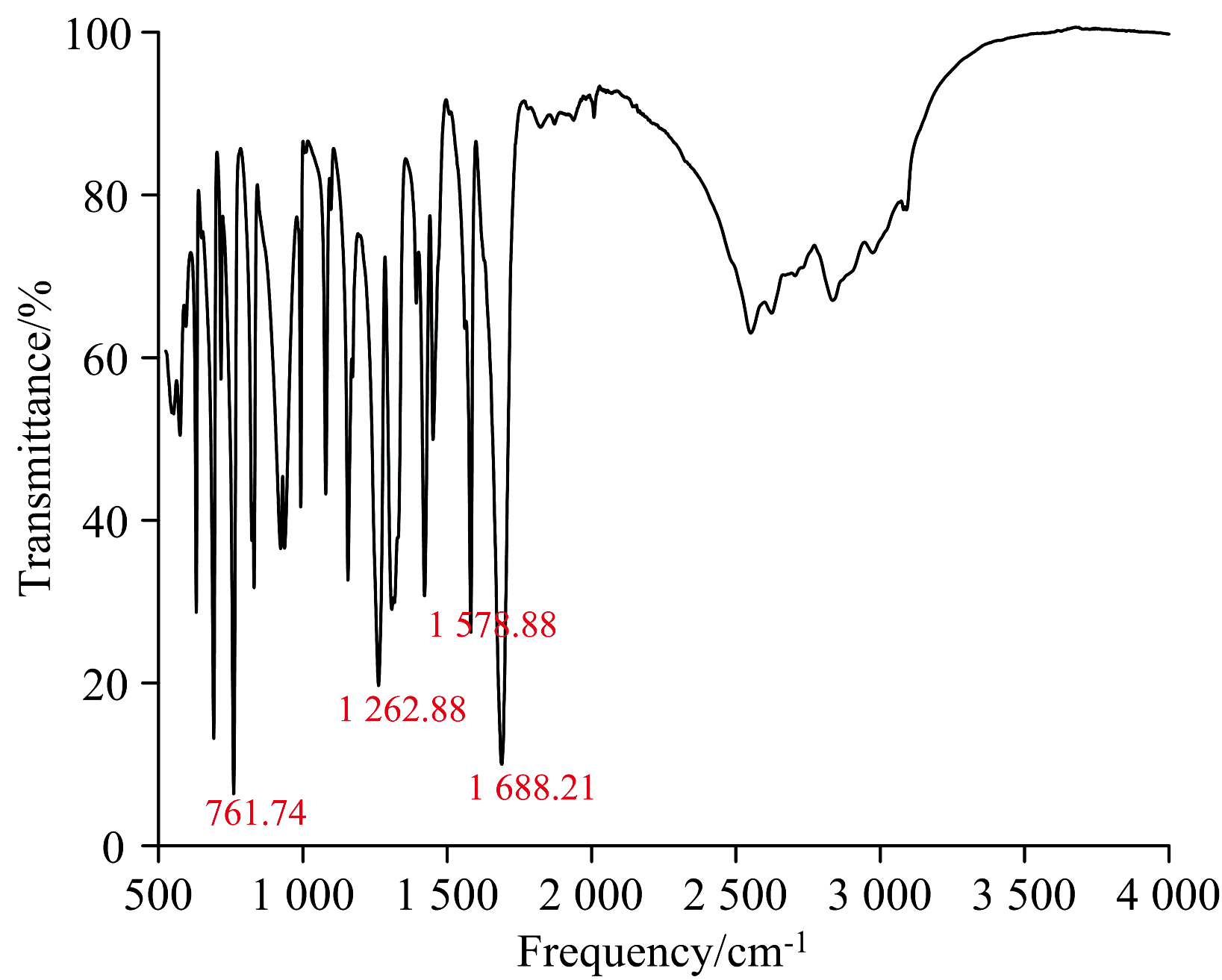 | 图2 实验得到的BP6DC的红外光谱Fig.2 The infrared spectrum of BP6DC obtained by experiment |
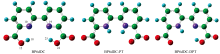
基于DFT/TD-DFT理论Cam-b3lyp/6-31G(d, p)[36, 37]基组优化了BP6DC在CYH, DCM和DMSO溶剂中的S0态和S1态的结构。 图3是BP6DC分子发生单质子转移(BP6DC-PT)和双质子转移(BP6DC-DPT)前后的结构变化图。 其中单质子转移指的是BP6DC沿O12— H18…N10或者O24— H25…N19方向发生质子转移, 双质子转移指的是BP6DC沿O12— H18…N10和O24— H25…N19同时发生质子转移。 为了比较分析表2列出了三种溶剂中的双氢键所涉及到的氢键的几何参数。 由于BP6DC分子本身就具有非常好的对称性, 所以在CYH溶剂中, BP6DC的两个氢键O12— H18…N10和O24— H25…N19同时得到增强, 相反在DCM和DMSO溶剂中两氢键的变化正好相反。 在CYH溶剂中O12— H18键在基态时键长为0.980 Å , 光激发跃迁到第一激发态时键长为0.992 Å 。 H18…N10键则发生相反的变化, 即基态时键长为1.965 Å , 激发态时键长缩短为1.897 Å 。 另外键角δ (O12— H18…N10)和δ (O24— H25…N19)在基态或激发态时并没有太多的变化。 由于分子内氢键的键长变化可以很好的反应光激发的影响, 即证明CYH溶剂中BP6DC的氢键在激发态下得到增强。 同样O24— H25和H25…N19键也具有相似的变化过程, 所以我们认为极性低的溶剂并没有使BP6DC的结构发生变化。 我们在观察DCM和DMSO溶剂中BP6DC分子的结构变化时, 发现O12— H18键从基态到激发态的键长分别拉长了0.016和0.003 Å , H18…N10键分别缩短了0.003和0.005 Å ; 虽然O12— H18键和H18…N10键的变化比CYH中的弱但是确定发生了类似的变化趋势, 同样在激发态下O12— H18键和H18…N10的氢键得到了增强。 然而O24— H25和H25…N19在DCM和DMSO溶剂中发生了完全相反的变化趋势。 比如O24— H25键在激发态时分别缩短到0.978和0.968 Å ; H25…N19键则拉长到2.312和2.320 Å ; 键角δ (O12— H18…N10)和δ (O24— H25…N19)也发生了比较明显的变化。 从O24— H25和H25…N19的键长变化中我们推测在这两种极性溶剂中BP6DC分子的对称性受到影响。 下面的振动光谱和弱相互作用的计算中将进行详细的分析。
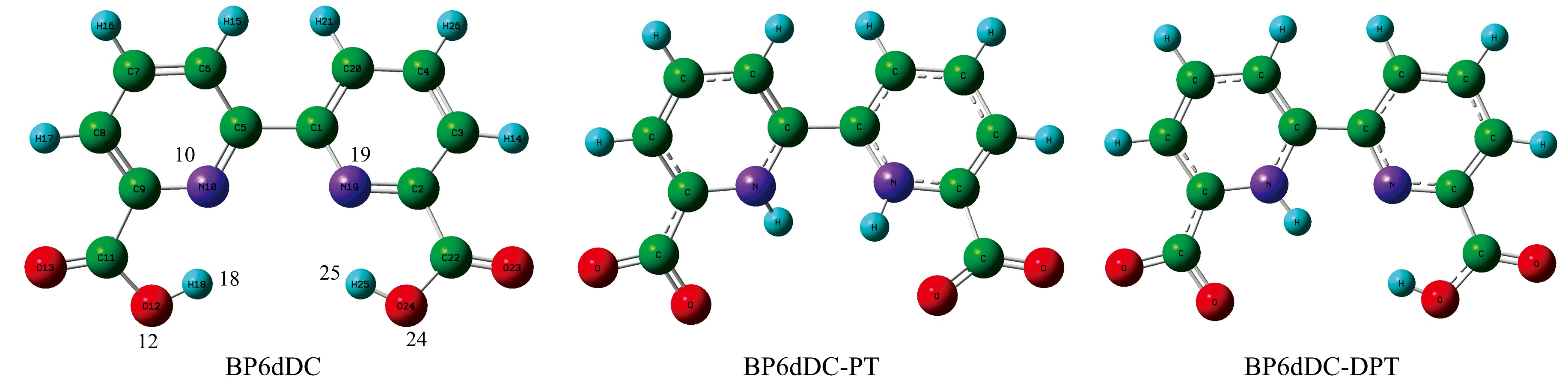 | 图3 BP6DC, BP6DC-PT和BP6DC-DPT结构 紫色: N; 红色: O; 蓝色: HFig.3 BP6DC, BP6DC-PT and BP6DC-DPT structures purple: N; red: O; blue: H |
| 表2 BP6DC在CYH, DCM和DMSO三种溶剂中的相关氢键几何参数 Table 2 The related hydrogen bond geometric parameters of BP6DC in CYH, DCM and DMSO solvents |
红外光谱的变化情况同样可以证明氢键的增强或减弱情况[38]。 因此在这一节中我们利用同样的方法模拟了BP6DC在CYH, DCM和DMSO溶剂中的红外光谱。 我们分别计算了S0态和S1态时的红外光谱, 观察到了与上一节的氢键参数一致的变化规律。 如图4所示, 在CYH溶剂中, S0态下的BP6DC在振动波数为3 600 cm-1处存在O12— H18和O24— H25同时的伸缩振动, 而S1态下在3 392 cm-1处出现O12— H18键单独的伸缩振动, 与S0态相比频率红移了208 cm-1。 同样在S1态下, O24— H25键在3 584 cm-1处出现了伸缩振动, 其频率与S0态相比仅仅红移了16 cm-1。 这表明, 在S1态下, CYH使BP6DC的氢键得到了增强。 在DCM溶剂中, 基态的BP6DC在3 636 cm-1处存在O12— H18 键和O24— H25键协同拉伸振动。 而在激发态下, 分成两个峰, 分别红移到3 571 cm-1和蓝移到3 823 cm-1处。 类似的, DMSO溶剂中, BP6DC分子在基态时, 振动波数为3 627 cm-1处也出现O12— H18键和O24— H25 键协同拉伸振动。 同样在S1态时被分成两个单独的峰, 振动波数分别红移到3 571 cm-1和蓝移到3 817 cm-1。 我们发现随着溶剂极性的增强, O12— H18键在S1态的伸缩振动的波数红移程度在减小, 如CYH(208 cm-1)→ DCM(65 cm-1)→ DMSO(56 cm-1)。 说明溶剂极性影响氢键在激发态下的增强程度。
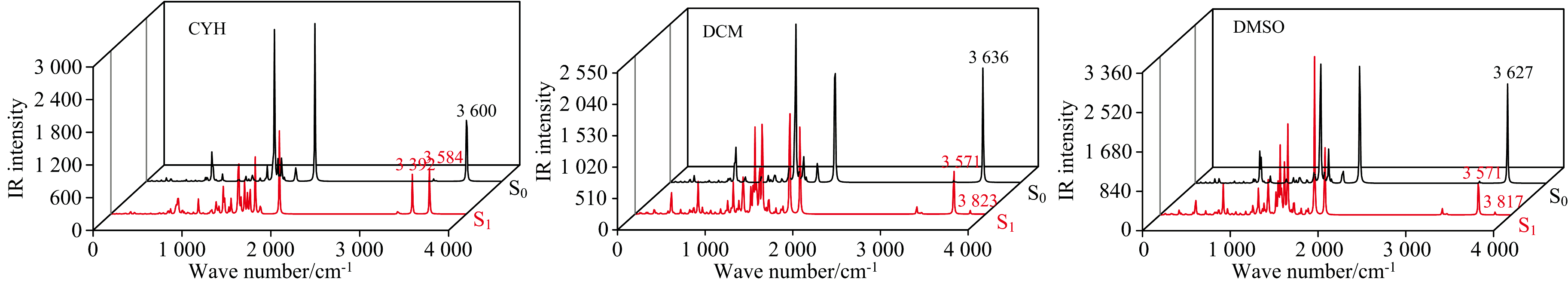 | 图4 BP6DC在CYH, DCM和DMSO中的IR光谱Fig.4 IR spectra of BP6DC in CYH, DCM and DMSO |
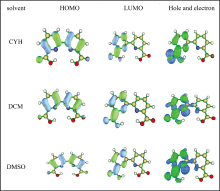
当BP6DC被激发到激发态后, 分子内电子会经历重新分布。 激发贡献主要来自于最高占据轨道(HOMO)到最低未占据轨道(LUMO)的分子轨道跃迁[39]。 在这一节中我们为了探索电荷分布特性, 表3中列出了BP6DC光诱导跃迁信息, 主要考虑最强吸收峰所对应的HOMO→ LUMO的跃迁占比。 单看HOMO→ LUMO的变换不难看出跃迁形式是π — π * , 另外值得注意的是O12原子的电子密度降低, N10原子的电子密度的增高。 然而只关注BP6DC分子在三种溶剂中HOMO→ LUMO的单个轨道跃迁是不可靠的, 我们认为BP6DC的跃迁过程包括多种轨道跃迁, 因此我们在图5中分别列出了HOMO, LUMO, 空穴-电子轨道分布图。 空穴-电子轨道分布可以非常直观的看出分子内电子的分布情况, 除此之外, 通过电子转移距离(D)和空穴-电子的分离程度Sr(r)判断出激发类型。 对空穴-电子Sr(r)分布的重叠部分进行积分, 以确定空穴和电子的空间间隔, Sr(r)函数表示为
$S_{r}(r)=\sqrt{\rho^{\text {hole }}(r) \rho^{\text {ele }}(r)}$(1)
式(1)中, ρ (r)hole和
| 表3 BP6DC在CYH, DCM和DMSO溶剂中的激发波长λ , 振子强度f Table 3 Excitation λ , oscillator strength f of BP6DC in CYH, DCM and DMSO solvents |
 | 图5 BP6DC在CYH, DCM和DMSO溶剂中的HOMO和LUMO轨道以及Hole-electron图 在Hole-electron中, 蓝色代表空穴分布, 绿色代表电子分布Fig.5 HOMO and LUMO orbitals and Hole-electron diagrams of BP6DC in CYH, DCM and DMSO solvents In the Hole-electron diagrams, blue represents the hole distribution, and green represents the electron distribution |
| 表4 PB6DC在CYH, DCM和DMSO溶剂中的空穴和电子质心距离(D), 空穴-电子的重叠函数(Sr)和空穴-电子分离函数(t) Table 4 Hole-electron centroid distance (D), hole-electron overlap function (Sr) and hole-electron separation function (t) of PB6DC in CYH, DCM and DMSO solvents |
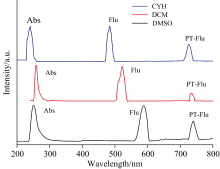
测定了BP6DC在低极性CYH, 中极性DCM和极性较大的溶剂DMSO中的稳态吸收光谱与荧光光谱, 如图6所示。 BP6DC在三种溶剂中归一化的吸收峰的位置分别在247、 260和240 nm, 在三种溶剂中吸收峰的位置没有明显的差异。 实验所得的结果与计算所得的电子吸收谱非常吻合, 说明我们所选的基组是比较准确的。 测量荧光光谱时我们发现了两个尖峰, 先前的研究表明这两个峰中一个是烯醇态发射出来的, 另一个峰则是酮态发射出来的。 如图6所示, 在CYH溶剂中, 247 nm是测得的吸收峰, 585 nm处的发射峰是BP6DC的烯醇式构型的荧光峰, 740 nm的荧光峰则是来自于BP6DC-PT(酮式构型)。 另外可以发现BP6DC的烯醇式和酮式构型都发生了斯托克斯位移。 因此我们有理由推测, 烯醇式BP6DC通过吸收光子发生ESIPT过程, 转化为相应的BP6DC-PT。 其余两种溶剂也出现类似的现象, 他们出现的两个发射峰同样来自于BP6DC和BP6DC-PT转化过程中存在的较大的斯托克斯位移。 最后我们发现随着溶剂极性的增强, BP6DC-PT发射峰出现蓝移。 到此我们证实BP6DC的ESIPT机制可以通过溶剂极性来调控。
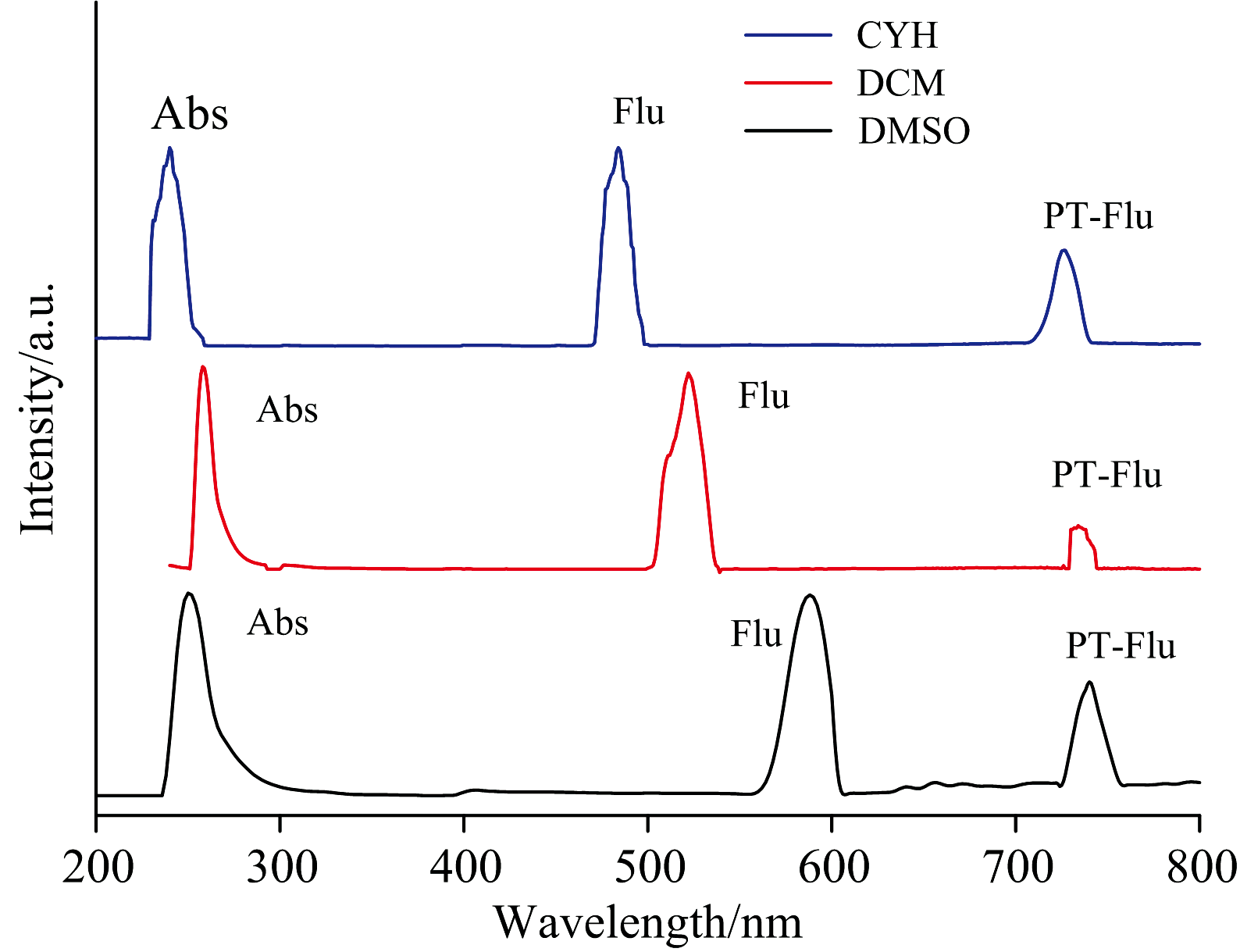 | 图6 测定BP6DC在CYH, DCM和DMSO溶剂中吸收光谱和荧光光谱Fig.6 Measured absorption and fluorescence spectra of BP6DC in CYH, DCM and DMSO solvents |
为了进一步推进真实空间中的分子内氢键相互作用, 利用了文献[40]提出的一种能够在真实空间中想象非共价相互作用的方法。 这种方法可以可视化研究弱相互作用, 通过降低密度梯度(RDG)和电子密度ρ (r)准确的表示不同类型的相互作用与其强度。 RDG函数可以通过填充颜色的方法来区分非共价键相互作用, 其表达式为
式(2)和(3)中, ρ (r)表示弱相互作用的反应强度, Sign[λ 2(r)]则表示反应的类型。 当两式相乘, 即Sign[λ 2(r)]ρ (r), 表示投影到RDG等值面上时, 弱相互作用的位置。 从键合类型方面考虑, λ 2(r)> 0, 表示共价键; λ 2(r)< 0, 表示非共价键。 当Ω (r)为负值时, 表示存在吸引力相互作用, 如氢键相互作用; 而Ω (r)为正值时, 则表示存在排斥相互作用。 当Ω (r)为0时, 表示存在范德华相互作用。 我们利用Multiwfn 程序, 将轮廓值设置为0.05, 将RDG值设置为-0.04~0.02。
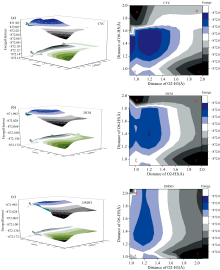
图7所示是处于S1态的BP6DC分子在CYH, DCM和DMSO三种不同极性的溶剂中的散点图和填色等值面图, 蓝色区域表示强吸引作用(氢键, 强卤键等), 绿色区域表示范德华作用力, 红色区域表示互斥作用(环, 笼中的位阻效应)。 如图5所示, BP6DC在CYH溶剂中的两个峰值分别出现在-0.035(O12— H18…N10)和-0.031(O24— H25…N19)处。 两个峰都说明O12— H18…N10和O24— H25…N19都存在强吸引作用, 即氢键。 相反, 在DCM和DMSO溶剂中, 仅在O12— H18…N10处出现强吸引作用, 分别在-0.028和-0.026。 另外随着溶剂极性的增强, 吸引作用即氢键的强度在减弱。 例如CYH(-0.035和-0.031)< DCM(-0.028)< DMSO(-0.026)。 到此我们进一步认为ESIPT过程可以通过溶剂极性来调控。
 | 图7 BP6DC分子在CYH, DCM和DMSO溶剂中约化密度梯度与Ω (r)的分布Fig.7 Distribution of reduced density gradient and Ω (r) of BP6DC molecule in CYH, DCM and DMSO solvents |
为了更清楚的解释BP6DC的激发态分子内质子转移机制, 首先我们计算了基态和第一激发态的BP6DC分子在三种溶剂中的势能曲面。 以0.1 Å 为步长, 对处于S0和S1态的BP6DC分子在CYH, DCM和DMSO三种溶剂中的O12— H18和O24— H25键从0.9到2.1进行同时的扫描, 如图8所示。 在三种溶剂中, S0态的BP6DC分子的势能随着O12— H18和O24— H25键的变长而增加。 因此无法在S0态下自发的发生质子转移。 从S1态所对应的等值面图可以观察到三种溶剂下都存在A、 B、 C和D四个势能极低点, 分别对应以下构型, A对应BP6DC构型, B与C对应BP6DC-SPT构型, D是BP6DC-DPT构型。 表5所示是CYH, DCM和DMSO溶剂中四个势能最低点的位置与势能大小。
 | 图8 PB6DC分子在三种溶剂下S0, S1态的势能曲面及S1态的势能等值面Fig.8 Potential energy surface of S0, S1 states and potential energy isosurface of S1 state of PB6DC molecule in three solvents |
| 表5 通过扫描BP6DC在S1态下O12— H18和O24— H25键所得到的四个势能最低点 Table 5 Four lowest potential energy points obtained by scanning the O12— H18 and O24— H25 bonds of BP6DC in the S1 state |
从表5中可得在CYH溶剂中B点与C点有相等的能量最低点, 图8(a)的等值面图中也能够观察到相似的结果。 相反在DCM和DMSO溶剂中B点和C点的能量不同, 说明DCM和DMSO两种极性溶剂中分子的对称性受到影响。 计算了三种溶剂中从A→ D点势垒如下: 在CYH溶剂中44.67 Kcal· mol-1, DCM溶剂中40.83 Kcal· mol-1, DMSO溶剂中是41.07 Kcal· mol-1, 由以上数据可得BP6DC在三种溶剂中都无法同时完成激发态分子内双质子转移过程。
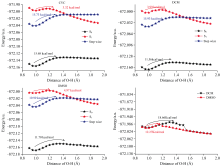
分别以0.05 Å 为步长, 扫描了处于S0, S1态的BP6DC分子, 在三种溶剂中的O12— H18键和S1态下完成H18的转移后的O24— H25键的势能曲线, 如图9所示。 通过扫描S0态时O12— H18的势能曲线发现在CYH, DCM和DMSO溶剂中的势垒分别为15.68、 11.56和11.70 Kcal· mol-1, 同样无法完成质子转移过程。 扫描S1态O12— H18键的势能曲线发现克服的势垒分别为3.23、 3.99和4.97 Kcal· mol-1均可以发生ESIPT。 另外需要注意的是ESIPT路径的势垒随着溶剂极性的增强而增加, 这意味着非极性溶剂可能有助于让BP6DC分子发生ESIPT过程。 为了验证是否发生逐步的激发态分子内双质子转移过程, 以同样的方法计算了B, C→ D过程的势垒, 发现三种溶剂下都存在较高的势垒15.73、 15.93和19.99 Kcal· mol-1, 因此同样无法完成逐步的激发态双质子转移过程。
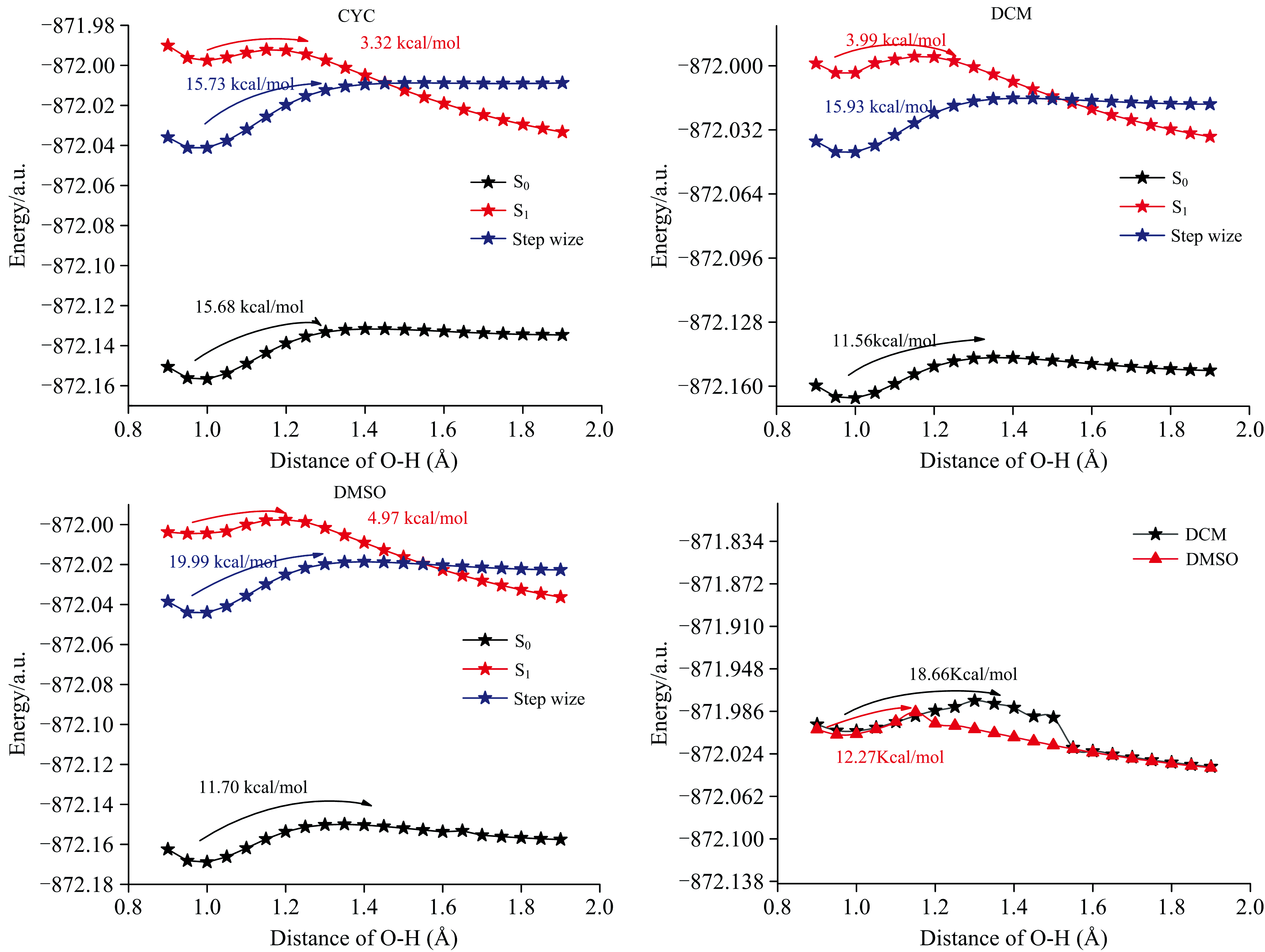 | 图9 BP6DC分子在三种溶剂下S0, S1态扫描O12— H18键的势能曲线Fig.9 The potential energy curves of BP6DC molecule scanning O12— H18 bond in S0 and S1 states in three solvents |
我们使用相同的计算方法和步骤增量对O24— H25键的势垒进行了扫描。 结果显示, 二氯甲烷(DCM)的势垒高度为12.27 Kcal· mol-1, 二甲基亚砜(DMSO)的势垒高度为18.66 Kcal· mol-1。 基于这些发现, 我们得出结论, 在DCM和DMSO溶剂中, 单向的ESIPT是可行的。 这一结论强调了溶剂对激发态质子转移过程的方向性和可行性的影响。
对BP6DC分子在一系列极性和非极性溶剂中的吸收和荧光光谱进行了实验性调查: 环己烷(CYH)、 二氯甲烷(DCM)和二甲基亚砜(DMSO)。 我们的发现表明, BP6DC在每个溶剂中都表现出显著的斯托克斯位移, 这表明溶剂的极性性质对BP6DC的分子对称性造成破坏, 如在CYH溶剂中, BP6DC的两个氢键O12— H18…N10和O24— H25…N19同时得到增强, 相反在DCM和DMSO溶剂中两氢键的变化正好相反, 因此影响了他们的质子转移过程。 基于这些观察, 进一步进行了控制分子内双氢键、 空穴-电子分布和激发态分子内质子转移行为的理论研究。 我们对氢键几何结构和红外振动光谱的分析表明, 所有溶剂在S1激发态中氢键都有所增强, 增强程度按照CYH> DCM> DMSO的顺序排列。 值得注意的是, 前线轨道(MOs)分析突出了N10的氢键接受能力。 通过检查空穴-电子轨道, 观察到BP6DC分子在CYH中经历了电荷转移激发, 而DCM和DMSO环境则表现出局部激发的特征。 这种区别对于光激发捕获位置H18和H25的羟基质子至关重要。 此外, 势能分析提供了明确的证据, 表明H18和H25质子都可以在CYH溶剂中进行激发态质子转移。 相反, 在极性溶剂DCM和DMSO中, 由于分子对称性被破坏, 只有H18质子可以转移。 这项工作不仅阐明了BP6DC的复杂激发态动力学, 而且还介绍了一种溶剂极性可以调节这些现象的机制。 这些见解将增强我们对BP6DC光物理性质的理解, 并可能促进荧光材料开发的进步。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|