{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
原位红外光谱技术在生物质转化研究中的应用
[卢思 , 陈晓丽, 梁正, 王小曼, 苏秋成, 亓伟, 付娟
, 陈晓丽, 梁正, 王小曼, 苏秋成, 亓伟, 付娟* ]
, 陈晓丽, 梁正, 王小曼, 苏秋成, 亓伟, 付娟]
|
|


作者简介: 卢 思, 女, 1994年生, 中国科学院广州能源研究所分析测试中心工程师 e-mail: lusi@ms.giec.ac.cn
生物质是地球上最丰富的生物可再生资源, 已被认为是替代化石资源缓解全球能源和环境危机的潜在能源。 作为碳的唯一可再生来源, 生物质可通过多种催化手段和转化途径转化为各种高价值化学品和能源密集型生物燃料。 在生物质的转化利用过程中, 分析反应物、 中间体、 副产物和产物的结构和组成等信息对于深入研究反应路径和反应机制, 提高转化效率至关重要。 具有高灵敏度和实时性的原位红外光谱技术, 是基于傅里叶红外光谱的原理, 通过装配原位反应池来在线监测实际条件下进行的化学反应, 获得物质在反应过程中随反应条件而变化的特征谱图, 在生物质转化的分析研究中具有广泛的应用前景。 本文综述了近年来原位红外光谱技术在生物质转化研究中的应用, 介绍了利用原位红外光谱技术表征分析生物质的转化机理, 表征手段包含透射、 漫反射(DRIFTS)和衰减全反射(ATR)等模式。 针对生物质高价值利用的不同转化策略进行了分类综述, 重点讨论了原位红外光谱技术在研究热解反应、 化学催化反应、 电催化反应机理等方面的最新进展, 反应过程涉及氢解、 加氢、 氧化、 还原、 异构化等多种催化手段, 通过跟踪反应物结构和官能团的变化, 深入研究反应路线、 反应机制和反应动力学。 并总结了原位红外光谱技术不同的测试模式分别在研究热解反应、 化学催化反应、 电催化反应机理时的优势和局限。 最后, 针对目前原位红外光谱技术应用于生物质转化过程中存在的一些挑战和困难, 展望了原位红外光谱技术的发展方向和潜力, 包括对原位池进行技术优化以提升极端反应条件的适配性和发展原位红外光谱与质谱、 拉曼光谱的多模态联用技术, 为深入研究生物质转化机理带来更为积极的影响。
Biomass, as the most abundant bio-renewable resource on Earth, has been recognized as a potential energy source to replace fossil resources and mitigate global energy and environmental crises. As the sole renewable carbon source, biomass can be converted into high-value chemicals and energy-intensive biofuels through various catalytic methods and conversion pathways. In the process of biomass conversion, it is essential to analyze the structure and composition of reactants, intermediates, by products, and products for the study of reaction pathways and reaction mechanisms to improve conversion efficiency. Based on the principle of Fourier infrared spectroscopy, in situ infrared spectroscopy technology with high sensitivity and real-time capabilities is used to monitor online chemical reactions by assembling an in situ reaction tank. It is capable of obtaining the characteristic spectra of substances changing with the reaction conditions, and has a wide range of application prospects in the research of biomass conversion. In this paper, the application of in situ infrared spectroscopy in recent years to research biomass conversion is reviewed. The transformation mechanism of biomass is analyzed using in situ infrared spectroscopy, which includes transmission, diffuse reflection (DRIFTS), and attenuated total reflection (ATR) modes. A categorized review of various conversion strategies for the high-value utilization of biomass is presented, focusing on recent advances in in situ infrared spectroscopy in the study of pyrolysis, chemical catalysis, and electrocatalysis reaction mechanisms. The reaction process involves a variety of catalytic methods, including hydrolysis, hydrogenation, oxidation, reduction, and isomerization, among others. The reaction route, reaction mechanism, and reaction kinetics are studied in depth by tracking the changes in reactant structure and functional groups. It also summarizes the advantages and limitations of various testing modes in in situ infrared spectroscopy when applied to investigate the reaction mechanisms of pyrolysis, chemical catalysis, and electrocatalysis. Finally, addressing the current challenges and difficulties in applying in situ infrared spectroscopy to biomass conversion processes, this study proposes the future directions and potential of this technology. These include technical optimization of in situ reaction cells to enhance compatibility with extreme reaction conditions, as well as the development of multimodal coupling techniques, such as integrating in situ infrared spectroscopy with mass spectrometry and Raman spectroscopy. Such advancements are expected to provide deeper insights into biomass conversion mechanisms and foster more significant breakthroughs in this field.
由于不可再生化石资源的迅速开发和枯竭, 能源短缺和环境恶化已成为世界性问题[1]。 生物质作为碳的唯一可再生来源, 已被证明具有升级为有价值的衍生化学品和生物燃料的巨大潜力[2, 3]。 近年来各国政府宣布了在21世纪下半叶实现碳中和的国家战略, 进一步促进了生物质在可再生能源领域的快速发展[4]。 生物质成分复杂, 主要由40%~50%的纤维素、 25%~35%的半纤维素和15%~20%的木质素构成[5, 6]。 其中纤维素和半纤维素由d-葡萄糖吡喃糖单元组成, 可以进一步升级为乙酰丙酸、 5-羟甲基糠醛、 糠醛、 甲酸等多种平台化合物, 是生产精细化学品和燃料的理想起始原料[7]。 为了实现生物质的高价值利用, 研究者们开发了包括热化学法、 多相催化、 电催化法等转化策略来获得生物燃料和衍生化学品[8, 9, 10]。
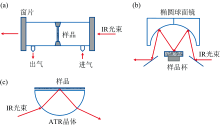
在生物质的高值化转化利用过程中, 表征分析反应物、 中间体、 副产物和产物的结构和组成等信息对于深入研究反应过程, 提高目标产物的转化效率至关重要[11, 12]。 近年来, 原位红外光谱技术(FTIR)在表征生物质转化研究中获得了广泛的应用。 实时在线监测的原位红外光谱技术, 是基于傅里叶红外光谱的原理, 通过装配原位反应池来分析实际条件下进行的化学反应[13]。 原位红外光谱技术通过简单的样品处理, 模拟实际反应环境, 实时原位监测样品或反应体系随反应条件的变化而变化的信息, 从而总结发现其中规律, 通过跟踪这些物质结构和官能团的变化, 深入研究反应路线、 反应机制和反应动力学[14, 15]。 根据不同反应特点, 发展了原位透射、 原位漫反射(DRIFTS)和原位衰减全反射(ATR)等模式[16], 示意图如图1所示。 这三种测试模式具有不同的工作原理, 根据样品形态、 检测需求和研究目标等不同特征, 对其优缺点和适用场景进行了总结和比较, 如表1所示。
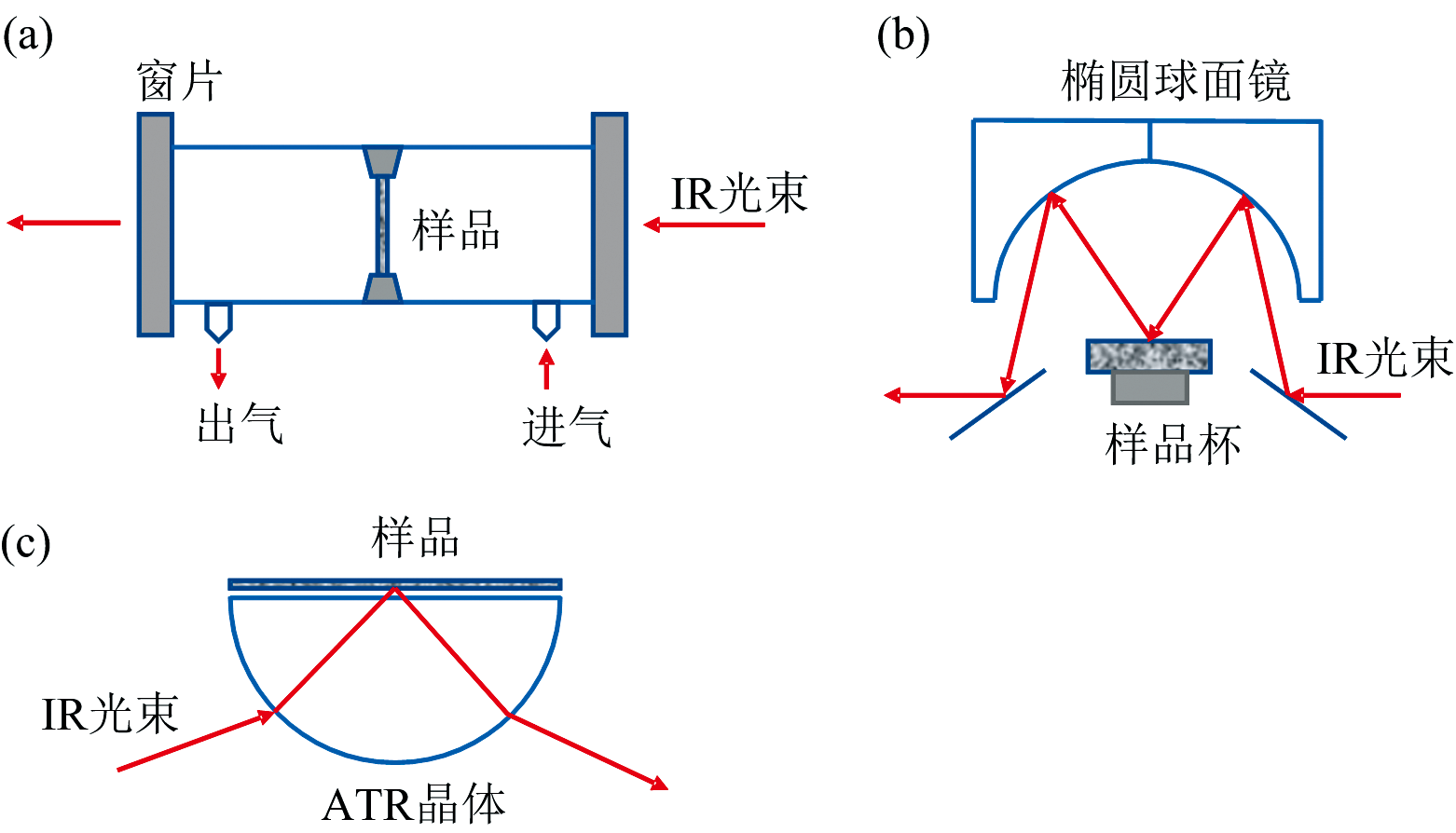 | 图1 原位FTIR测试模式示意图 |
| 表1 原位FTIR三种模式的介绍与对比 Table 1 Introduction and comparison of the three modes of in situ FTIR |
随着生物质资源高值化转化的深入研究, 揭示反应过程中分子键合演变与中间体动态行为已成为机理研究的核心挑战。 不同模式的原位红外光谱技术因具备实时监测能力、 原位反应兼容性及化学指纹特异性, 在生物质转化机理解析中展现出独特优势。 本文聚焦原位红外光谱技术在生物质转化研究中的应用进展, 包括热解反应、 化学催化反应、 电催化反应等机理研究及其他前沿应用, 旨在为生物质转化研究提供原位实验和谱学方法参考。
热解是一种“ 无焰” 的生物质热化学转化技术, 通过热解可以将生物质转化为环境友好且具有实际价值的低碳生物燃料、 材料和化学品[23]。 显然, 热解技术的优化和改进对废弃生物质的资源化利用和低碳能源社会的建设具有重要意义[24]。 生物质热解过程非常复杂, 涉及传热、 多组分化学反应、 相变、 传质等过程, 且生物质热解效率和产物会受到温度、 升温速率和保温时间等外部因素的影响[25, 26]。 对生物质热解机理进行基础性研究, 有助于分析生物质热解过程, 促进生物质热解技术的发展[27, 28]。
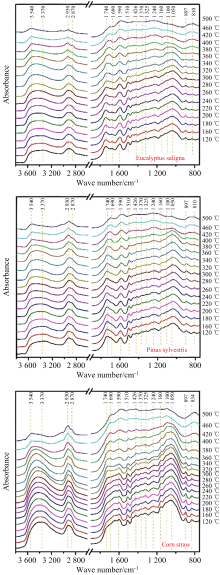
采用原位红外光谱技术可以对生物质热解过程中的分解产物、 分子演化以及多组分在分子水平上的相互作用等进行深入研究[29, 30, 31, 32]。 Wang等利用原位FTIR漫反射技术监测生物质热解过程中各种特征结构的演变, 结果如图2所示。 通过对比盐桉、 山松和玉米秸秆三种生物质的热解机制发现, 同一官能团在不同生物质样品中的热稳定性并不完全相同, 生物质的结构差异会导致不同的热解行为。 进一步结合热解路径的密度泛函理论(DFT)计算证明, 软木半纤维素中葡甘露聚糖的糖苷键的断裂和O-乙酰侧链的解离比硬木和秸秆更容易发生, 木聚糖中α -醚键的热稳定性强于葡甘露聚糖, 其断裂发生在较高的热解温度下[33]。 Dai等利用原位FTIR漫反射技术, 对半纤维素热解过程中的结构演化进行了研究, 对比了三种半纤维素模型化合物(木糖、 木糖二糖和木聚糖)的热解机理, 结果表明木聚糖热解初期以侧链解离为主, 其次是糖苷键的裂解和木吡喃糖环的打开, 侧链较多的木聚糖比木糖和木糖二糖更容易通过热解环断裂反应生成小分子化合物[34]。
 | 图2 不同热解温度下盐桉、 山松和玉米秸秆的原位漫反射光谱图[33]Fig.2 In-situ DRIFT spectra of eucalyptus saligna, pinussylvestris and corn straw at different pyrolysis temperatures |
Liu等采用原位FTIR对花生壳基质中官能团的热解机理进行了研究, 创新地引入原位FTIR去卷积技术, 对花生壳热解过程中固体基质官能团的实时演化和热动力学进行了识别和量化, 阐述了总OH基团在20~380 ℃、 脂肪族C— Hn基团在20~500 ℃、 C=O基团在260~500 ℃、 C— O基团在300~500 ℃过程中的多阶段热解动力学, 提出了反应机理为扩散反应, 热反应途径以传热和传质为主[35]。 Xie等采用原位FTIR分析了玉米秸秆与低密度聚乙烯共热解过程中挥发性产物的组成和主要官能团的变化, 发现官能团在固体基质上的反应活性依次为OH> C— O> C— H> C=O> C=C。 通过进一步分析揭示了共热解过程中形成生物炭的两种可能途径, 第一种途径涉及长链脂肪烃的环化和随后的聚合; 第二种途径是将烯烃、 醇、 醛和其他小分子聚合成前体, 在二次聚合过程中形成聚合物, 最终形成生物炭[36]。
在生物质热解反应的原位FTIR分析中, 通常采用透射模式或漫反射模式, 各有其独特的优缺点。 透射模式能够检测到热解过程中产生的低浓度气体(如CO、 CO2、 CH4等)和挥发性有机物, 具有高灵敏度和定量分析能力, 但生物质热解过程中可能生成炭或焦油等不透明产物, 透射模式难以穿透这些物质, 对固体残留物的分析能力有限。 漫反射模式可直接分析原始生物质颗粒及其热解残留物, 但由于光散射效应, 漫反射模式的信噪比通常较低, 可能影响对低浓度气体或微弱信号的检测。 总之, 通过原位FTIR技术分析生物质在热解过程中结构的变化, 可以初步识别热解过程中生成的产物和相应的反应路径, 然后可结合密度泛函理论等计算化学方法进一步探索生物质的结构演化, 深入分析特征结构和反应路径, 为生物质热解机理提供有价值的参考[37, 38, 39]。
为了经济地实现环境友好型的生物质精炼, 近年来研究者们探索了包括氢解、 加氢、 氧化、 异构化等多种催化方法将生物质转化为各种高价值化学品和能源密集型生物燃料[40, 41, 42]。 采用化学催化策略时通常包括两个步骤: 首先将生物质转化为平台分子, 然后通过催化剂将所得平台分子升级为有价值的化学品和生物燃料[43, 44]。 为了提高催化效率和转化率, 对反应过程中生成的产物、 中间体以及催化机理的研究至关重要, 而原位红外光谱技术是实时监测反应过程中吸附物种变化的最有效的手段之一[45, 46, 47]。
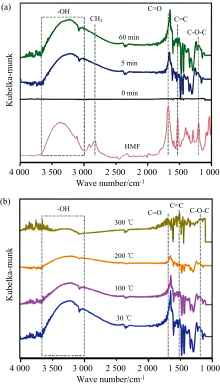
5-羟甲基糠醛(HMF)是一种重要的生物质衍生平台化合物, 可用于生产可持续生物产品、 生物燃料和聚酯[48]。 HMF中C=O基团的碳原子已被证明是分子中最亲电的中心, 因此容易被氢化[49]。 Dong等利用原位FTIR漫反射技术对锐钛矿负载金催化剂(Au/a-TiO2)催化5-羟甲基糠醛(HMF)选择性生成5-甲基糠醛(MF)进行了分析, 如图3所示, HMF的— OH在Au/a-TiO2上吸附的伸缩振动峰发生了红移, 表明HMF主要通过— OH吸附在催化剂表面, 有利于— OH的活化, 从而促进C— OH的断裂。 同时, 考察了不同温度下Au/a-TiO2对HMF解吸状态, 随着解吸温度的升高, 在3 000~3 600 cm-1处的— OH伸缩振动和1 668 cm-1处的C=O拉伸振动的强度逐渐降低, 但在200 ℃时仍能检测到, 表明HMF在Au/a-TiO2上的强烈吸附, 避免醛基的加氢[50]。
 | 图3 (a) Au/a-TiO2对HMF吸附的原位FTIR谱图和固体FTIR谱图; (b)不同温度下Au/a-TiO2对HMF解吸的原位FTIR谱图[50]Fig.3 (a) In situ FTIR spectra for Au/a-TiO2 on adsorption of HMF and the FTIR spectra for solid HMF; (b) In situ FTIR spectra for Au/a-TiO2 on desorption of HMF at different temperatures |
通过HMF中的C— OH直接选择性氢解生成MF仍然具有很大挑战性, HMF的氢解过程会形成各种中间产物, 因为其与氧有关的结构多种多样, 包括C— OH、 C=O和呋喃环, 导致最终的选择性不高, 例如HMF在Au/a-TiO2上的氢解为MF的收率为83.1%[50]。 为了应对这一挑战, Yu等制备了一种非贵金属催化剂(Ni-TT-Nb2O5), 可将HMF氢解为MF的选择性提高到99%。 采用原位FTIR分析了HMF在Ni-TT-Nb2O5表面的吸附方式, 发现随着反应时间的延长, C=O的峰会发生红移, 而C— OH的峰在整个反应过程中没有明显的位移, 这表明Ni-TT-Nb2O5表面吸附了C=O结构。 另外, 在升温解吸过程中发现— OH和C=O峰的信号变弱, 但即使在200 ℃时仍能观察到— OH和C=O峰的信号, 这表明 Ni-TT-Nb2O5表面对HMF中C— OH和C=O结构的吸附很强。 结合DFT计算证明了HMF以平行吸附方式吸附在Ni-TT-Nb2O5表面, C— OH键更容易被激活[51]。
甲酸(FA)作为最简单的羧酸, 是一种有价值的化学物质和势能载体, 从可再生资源如生物质获得FA是一种有前途的替代方案[52, 53]。 Yan等通过降低配位数的策略开发了一种有缺陷的MnO2催化剂, 以增强各种多元醇/糖的催化氧化生成甲酸。 采用原位FTIR研究了多元醇在Mnδ +— OV对上氧化为甲酸的反应机理, 随着温度的升高甘油中伯羟基活化, 在1 370 cm-1处的ρ (OH)和1 440 cm-1处的δ (OH)归属于甘油醛, 在羟基自由基的攻击下, 形成的甘油醛迅速转化为CH2OHCHOHCHOOH, 在Mnδ +— OV结构上发生C— C键劈裂, 伯羟基的优先活化和C— C键的断裂促进了甲酸的生成[54]。
生物质多元醇在负载贵金属(Pt、 Pd、 Ru和Au)上直接水相氧化的绿色非均相反应引起了人们的广泛关注。 Yan等采用简单的水热策略设计了一种负载在羟基磷灰石(HAP)上的单原子Pt催化剂, 该催化剂(Pt1/HAP)在C2-C4多元醇选择性氧化成相应的伯羟基酸的过程中具有较高的选择性和优异的稳定性。 通过原位FTIR表征发现随着温度的升高, 甘油在1 660~1 640 cm-1的分子间氢键峰逐渐消失, 在高温(≥ 80 ℃)以C=O键峰为主, 证明甘油转化为羧酸产物。 同时, 属于伯羟基的特征峰(1 125~1 075 cm-1)向高波长移动, 表明甘油的伯羟基被活化, 而属于仲羟基(1 200~1 125 cm-1)活化的特征峰保持不变, 说明甘油主要是通过伯羟基被氧化为甘油酸[55]。
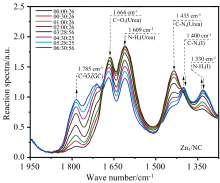
生物柴油产量的增长导致甘油过剩, 甘油羰基化生产有价值的碳酸甘油酯是有效利用甘油的一种化学方法[56, 57]。 Shi等采用湿法浸渍技术制备了碳基Zn单原子催化剂(Zn1/NC), 在温和条件下对甘油与尿素的羰基化表现出了优异的催化活性、 选择性和稳定性。 通过原位FTIR监测Zn1/NC催化剂上的羰基化反应过程, 结果如图4所示, 发现在反应初始阶段就可以观察到碳酸甘油酯和氨基甲酸酯中间体化合物的特征信号, 表明尿素可以很容易地被活化, 然后通过中间体与甘油反应生成碳酸甘油酯的产物[58]。 Wang等制备了一种B、 N、 P共掺杂的多孔碳材料, 在无金属条件下对甘油与尿素羰基化反应表现出优异的催化性能, 实现了高效的甘油转化和碳酸甘油酯选择性。 采用原位FTIR监测了甘油与尿素羰基化反应中官能团的变化, 随着反应的进行在1 326 cm-1处监测到新的吸收峰, 对应于关键反应中间体甘油聚氨酯的C— O伸缩振动。 当碳酸甘油酯产率缓慢增加时, 尿素的浓度仍显著降低, 这是主要用于副产物的生成, 且尿素会占据了一部分有限的活性位点, 抑制了中间体向目标产物的转化, 最终导致主要产物的选择性降低[59]。
 | 图4 原位FTIR监测Zn1/NC催化剂羰基化反应过程[58]Fig.4 Monitoring the carbonylation reaction course over Zn1/NC catalyst by in-situ FTIR |
5-羟甲基糠醛(HMF)因其含有C=O C— OH、 C=C等多个性质活泼的官能团, 通过催化加氢反应可转化为2, 5-二甲基呋喃(DMF)[60]。 Wang等通过层状双氢氧化物(LDHs)的还原制备了三种不同的Cu基多界面催化剂, 以控制HMF加氢的反应途径和产物选择性。 采用原位FTIR研究了DMF分子在Co@Cu/3CoAlOx和Co@Cu/5CoAlOx两个催化剂表面的吸附形式, 考察了关于C=C键加氢选择性的差异。 发现在前者中, DMF 中的共轭C=C— C=C结构以大π 键的形式吸附于铜颗粒表面, 而在后者中以C, C双原子形式吸附, 说明对C=C键作用更强。 考虑到Cu d轨道电子排布能对C原子产生强排斥作用, 以及两个催化剂的结构差异, 认为起吸附C=C键作用的是Cu— Co的共同作用[61]。
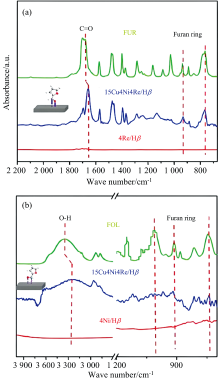
糠醛(FUR)一锅法直接加氢制2-甲基四氢呋喃(2-MTHF)具有设备要求低、 反应过程简单等优点[62]。 Peng等开发了一种三金属15Cu4Ni4Re/Hβ 催化剂, 以H2为氢源, 可高效地将FUR加氢脱氧转化为2-MTHF。 采用原位FTIR的ATR技术表征了FUR在催化剂上的吸附状态, 如图5(a)所示, 发现C=O基团峰红移至1 659 cm-1, 而呋喃环的峰位置保持不变, 说明只通过垂直构型吸附C=O基团, 并优先氢化成糠醇(FOL)。 同时FOL为底物进行原位红外吸附[图5(b)], 发现在催化剂表面OH基团的吸收峰明显红移至3 250 cm-1, 但呋喃环的吸收峰很弱, 说明该催化剂选择性吸附糠醇中的OH基团, 并优先将FOL转化为2-甲基呋喃(2-MF), 最后三种金属结合催化氢化生成2-MTHF[63]。
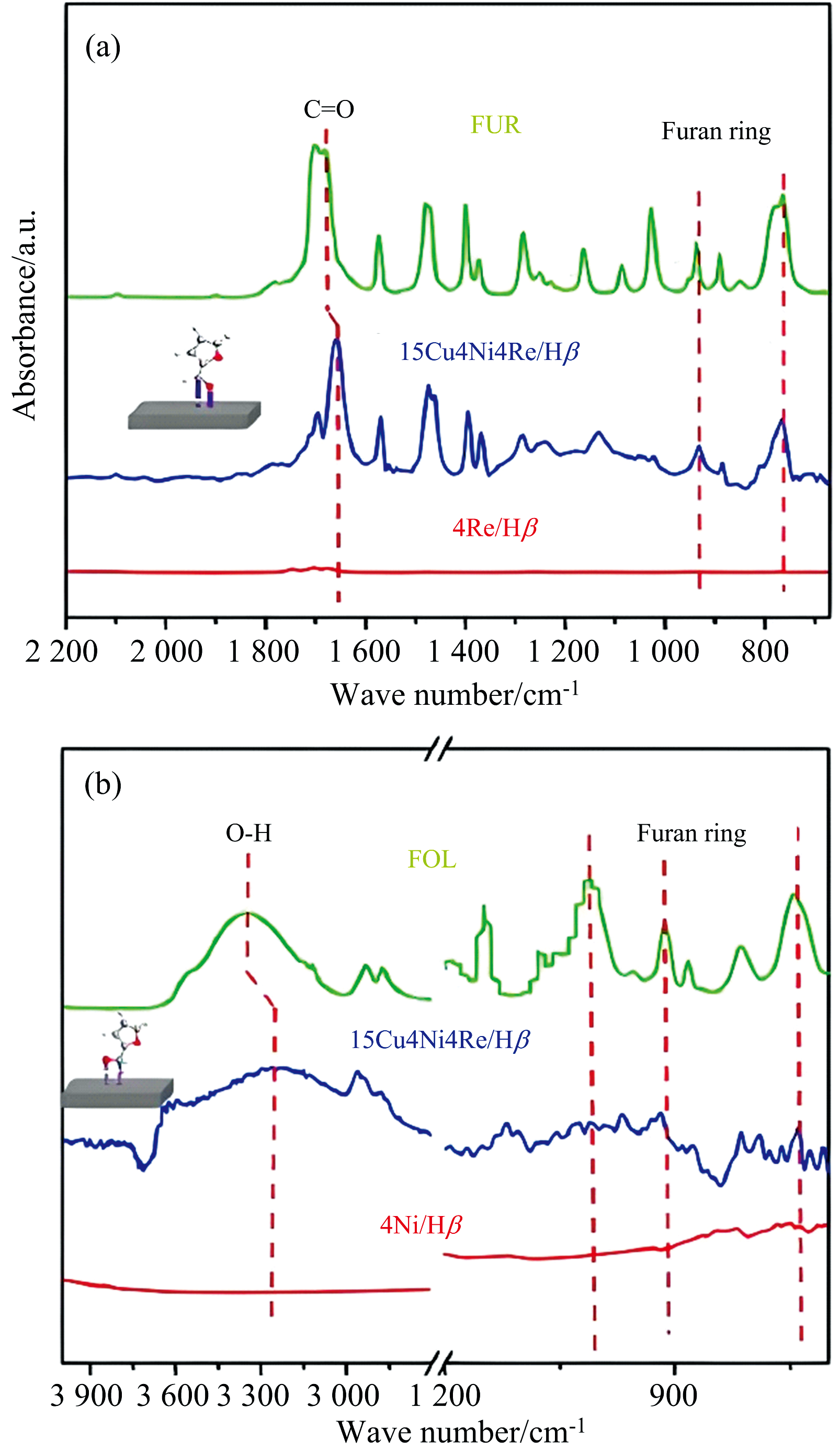 | 图5 在15Cu4Ni4Re/Hβ 催化剂上吸附FUR和FOL的原位FTIR-ATR谱图[63]Fig.5 In situ FTIR-ATR spectra of FUR and FOL adsorbed over 15Cu4Ni4Re/Hβ |
将生物质衍生物糠醛(FAL)经还原胺化制得糠胺(FAM)是合成胺类化学品的有效方法[64]。 Xue等通过在商用载体TiO2(P25)上负载超小尺寸的钌(Ru)团簇(~0.9 nm), 实现了光催化乙醇氢转移耦合生物质FAL还原胺化制备FAM的反应过程。 采用原位FTIR漫反射技术研究了FAL和FAM在0.1% Ru/P25和2% Ru/P25催化剂上的吸脱附行为, 红外光谱明显的红移现象表明Ru粒子的存在导致了FAL强的化学吸附。 当FAM吸附在催化剂表面时时, δ (N— H)的振动峰向较低波数移动(从1 620 cm-1移动到1 590 cm-1), 说明Ru物种有利于FAM中N— H键的化学吸附。 FAM吸附在2% Ru/P25表面上时, 1 440 cm-1处ν (C— N)的振动峰增强, 同时ν (C— N)振动峰从1 180 cm-1移动到1 150 cm-1处。 而在0.1% Ru/P25上的吸附时, 1 440 cm-1处没有观察到红外振动峰, 1 150 cm-1处的红外信号也非常弱。 这表明了产物FAM有利于在Ru团簇上脱附, 避免了副产物的形成[65]。 Xu等制备了一种MFI分子筛包覆Pt纳米粒子的Pt@Z5催化剂, 可以有效催化糠醛选择性直接还原胺化生成N-乙基-N-(呋喃-2-基甲基)乙胺。 采用原位FTIR对Pt@Z5上吸附的糠醛进行了分析, 并与催化剂Pt/Z5(在传统的Z5表面上负载Pt)进行了比较, 在3 700 cm-1附近的峰归因于沸石表面— OH振动的消耗, 表明沸石上的酸性位点与糠醛有强烈的相互作用。 对比糠醛的吸收峰发现Pt/Z5上吸附的糠醛明显弱于Pt@Z5, 但消耗的表面— OH基本一致, 表明对糠醛起吸附作用的主要是暴露的Pt原子, Pt@Z5暴露的Pt原子更多, 从而更好地吸附糠醛, 促进胺化过程中的转化[66]。
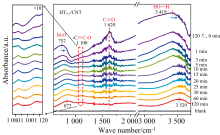
果糖作为食品甜味剂和生物精炼的核心成分其需求远大于葡萄糖, 可借助非均相催化的方式使高浓度葡萄糖高效异构化为果糖[67]。 Ye等采用自下而上的策略制备了镁铝水滑石/碳纳米管杂化催化剂(HT/CNT), 用于葡萄糖在水中异构化为果糖。 通过原位FTIR实验来监测中间体在反应过程中的演变, 如图6所示, 在120 ℃下, 在1 620 cm-1处检测到的吸收峰归属于葡萄糖的C=O振动, 这可以归因于第一步环葡萄糖开环。 当葡萄糖吸附在水滑石表面时, 在3 419和752 cm-1处的吸附峰分别为葡萄糖的OH振动和HT的M— O振动。 随着反应时间的延长, 葡萄糖和HT之间通过形成HO…H发生更多的相互作用, 氢键变得更加稳定, 在1 620 cm-1处C=O振动消失, 在1 100 cm-1处观察到烯醇结构的C=O振动, 说明反应过程中的同分异构过程。 吸收峰的变化表明HT的碱位置有利于去质子化过程, 从而产生烯二醇中间体, 并进一步转化为最终的环果糖[68]。
 | 图6 葡萄糖与HT/CNT在120 ℃下长时间反应的原位FTIR光谱[68]Fig.6 In-situ FTIR spectra during glucose reaction with HT/CNT at 120 ℃ with prolonged time |
近日, Sun等通过碱水热方法将铟(In)掺入单晶-纯二氧化硅沸石分子筛中形成边界骨架In和In2O3纳米颗粒(In-beta-TEAOH), 催化葡萄糖高效异构化的同时, 抑制了其在果糖的积累和降解过程中产生的副反应。 采用原位FTIR漫反射技术研究了不同量甲醇处理前后In-beta-TEAOH的羟基振动区的变化, 发现部分甲醇被分离的In原子吸附解离, 产生了一个比原来的In— OH更活泼的质子。 进一步增加甲醇的质量, 发现受甲醇刺激的活性质子在3 586 cm-1处的信号峰并没有随着甲醇质量的增加而增加, 与这个峰值相关的信号反而被减弱了, 说明过量的液体甲醇与In位点的相互作用抑制了与附近二氧化硅羟基连接的活性质子的产生, 这一发现为该反应体系在少量甲醇催化高浓度葡萄糖异构化的应用提供了有力的证据[69]。
在表征生物质化学催化反应过程方面, 原位透射模式能够检测催化转化反应中生成的低浓度气体和液体产物(如醇类、 酸类、 醛类), 具有高灵敏度的特点, 但对固体催化剂和不透明样品的分析能力有限; 原位漫反射模式可直接分析原始生物质、 平台化合物、 固体催化剂及其表面反应, 能够直接监测催化剂表面活性位点的变化和反应中间体的形成, 但对气态和液态产物的检测能力较弱; ATR模式对样品表面的化学变化非常敏感, 适合研究催化剂表面反应和界面化学, 能够直接监测液体反应物的化学变化, 特别适合研究水解、 氧化、 加氢等反应, 但ATR模式的探测深度通常为几微米, 难以分析体相反应或深层化学变化, 且不适用于气体分析。
近年来, 在生物质精炼中利用电催化技术取代传统苛刻的热催化无疑是实现绿色可持续发展的有效手段[70, 71]。 电催化过程是将电子从电极表面转移到底物, 通过在阴极和阳极分别发生还原和氧化反应将生物质衍生物转化为高附加值化学品。 与传统技术相比, 电催化具有常温常压温和条件、 反应效率高、 单位反应时间短、 易于扩大规模等独特优势[72, 73]。 电化学原位傅里叶变换红外光谱技术基于其指纹图谱和表面选择规则, 可以实时提供吸附物、 溶液种类以及参与电化学反应的中间体和产物种类的信息, 从而增强对界面过程的了解[74, 75]。
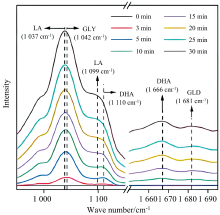
电催化氧化具有许多突出的优点, 如不需要化学氧化剂、 反应条件温和、 活性和选择性可控[76]。 Yan等开发了一种电催化策略, 通过在中等电位下使用氢氧化镍负载的金电催化剂(Au/Ni(OH)2), 实现了高电流密度下甘油和乙二醇分别升级为乳酸和乙醇酸的高选择性。 采用原位FTIR的电化学方法研究了Au/Ni(OH)2上甘油-乳酸的反应途径, 如图7所示, 反应3 min后观察到二羟基丙酮的信号(1 110和1 666 cm-1), 反应15 min后观察到低强度的甘油醛(1 680 cm-1)和乳酸(1 037和1 099 cm-1)的形成, 推断出Au/Ni(OH)2上的甘油酸转化为乳酸的过程包括甘油酸电氧化为二羟基丙酮(主要)和甘油醛(次要)[77]。 Chen等采用脉冲电位电解策略, 在铂基催化剂上将甘油选择性电催化氧化为甘油酸。 通过原位FTIR分析了甘油电氧化过程中表面微环境和吸附物种的变化, 发现与甘油氧化中间体相关的键的强度随着电势的增加和电解时间的增加而增加, 并且更高的电势会导致甘油氧化中间体的快速形成和积累。 同时发现反应过程中金属催化剂表面氢键呈增加趋势, 归因于负电荷有利于醇盐在催化剂表面的形成和吸附, 随着电势的增加, 表面OH-浓度也增加[78]。
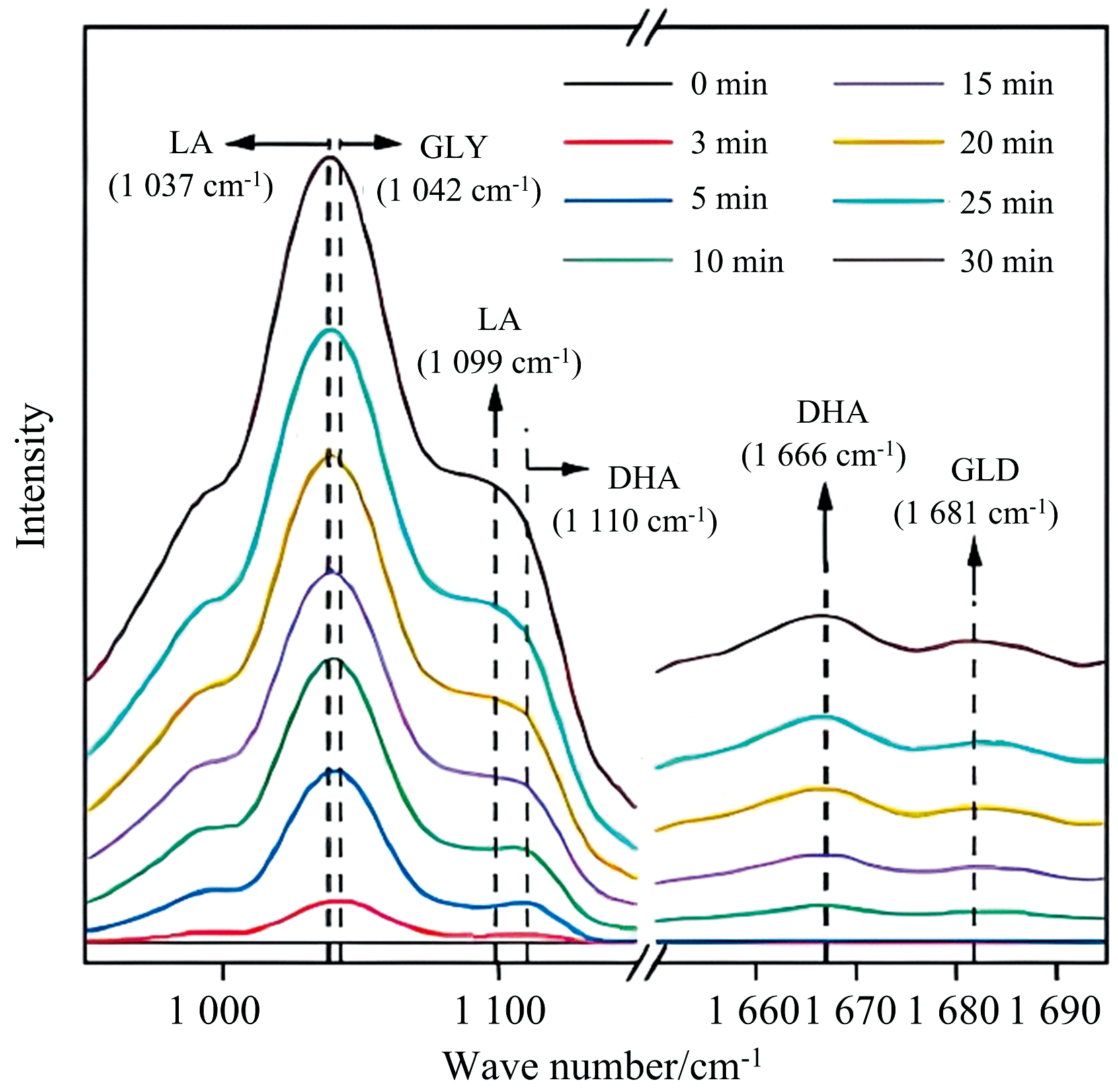 | 图7 甘油在Au/Ni(OH)2上氧化的原位FTIR谱图[77]Fig.7 In situ FTIR spectra recorded during glycerol oxidation on Au/Ni(OH)2 |
电催化还原以水作为氢源, 具有操作条件温和、 可耦合可再生电力驱动等特点[79]。 Wu等报道了在PdCu纳米珠线的催化下, 生物质衍生的丙酮酸(PA)和废硝酸盐(NO3-)通过电化学-化学-电化学一体化串联合成丙氨酸。 为了了解电化学偶联反应的途径, 采用原位FTIR的ATR-SERAS技术跟踪了PA和KNO3的在-0.3 VRHE条件下的实时反应过程, 发现随着反应时间的延长NO3-被电还原为* NH2OH中间体(1 472和1 560 cm-1), * NH2OH中间体与PA相互作用生成丙酮肟, 丙酮肟再被电还原为丙氨酸, 同时* NH2OH中间体与PA的偶联抑制了* NH2OH中间体还原为NH3[80]。 Zhang等采用电化学方法, 在简单合成的TiS2纳米片上将生物质衍生的呋喃醛选择性的进行还原胺化。 通过原位FTIR漫反射技术研究了层状TiS2纳米片在乙醇胺中对糠醛的电化学还原胺化(ERA)过程, 随着反应时间的增加, 在3 350 cm-1区域发现了— N— H与的拉伸振动, 同时发现了几个新形成的条带(1 557和1 508 cm-1), 这归因于5-(乙醇胺乙基)呋喃的— C— N拉伸振动, 另外在2 591 cm-1处的峰归因于有机分子与富含空位的TiS2纳米片之间的S— H• • • • C=O作用。 综合推断出醛基首先与乙醇胺发生亲核反应脱水生成亚胺, 然后在电子源的陪同下, 亚胺可以通过原位生成的H+快速还原为氨基呋喃[81]。
原位透射FTIR与电催化联用技术基于红外透射池同时作为电化学反应器, 集成光学窗口和三电极系统(工作电极、 对电极、 参比电极), 确保红外光穿透的同时进行电化学测试, 实时同步采集电化学信号与分子振动信息。 该联用技术时间分辨率高(毫秒级), 适合研究快速反应动力学, 且适用于液相体系, 如生物质水解液、 糖类电催化转化等。 但过程中水溶液干扰严重, 且电极设计受限, 可能影响催化性能。
原位漫反射FTIR与电化学联用技术采用三电极电解池, 集成红外光学窗口, 确保在施加电位/电流的同时采集表面光谱信号。 能够在生物质电催化转化过程中实时监测电极表面吸附物种、 中间产物及反应路径的动态变化, 为反应机理研究和催化剂优化提供关键信息。 但传统的漫反射电解池难以实现高电流密度下的稳定测试, 且不适合高含水量体系和气-液-固三相界面的精确控制。
ATR-SEIRAS联用技术通过金属纳米结构(如Au、 Ag)的局域表面等离激元共振效应, 将红外信号增强103~106倍, 显著提升检测灵敏度, 在监测原位反应过程中实现毫秒级时间分辨率, 捕捉瞬态中间体, 尤其适合生物质电催化中低浓度中间体的动态追踪。 但生物质分子(如木质素、 纤维素)通常具有高分子量和复杂结构, 在电极表面的扩散速率较慢, 导致ATR-SEIRAS检测到的信号可能仅反映表面吸附物种, 而非整体反应过程。 另外生物质转化常需高温或高压条件, 而ATR棱镜和密封材料可能失效, 需要结合实验条件设计更具兼容性的原位池体。
在生物质精炼过程中糖类转化是至关重要的一个环节, 分子筛催化剂的L酸能有效提高纤维素转化为葡萄糖的效率, 同时对于葡萄糖的异构化和转化也具有优异的催化性能。 Sun等将金属Ni通过离子交换掺入含有孤立金属Sn位点的纯硅Beta沸石中, 合成了一种优质的L酸催化剂。 采用吡啶作为探针分子, 通过原位FTIR透射技术表征了该催化剂酸的类型和相对含量等重要参数, 为改性催化剂酸量的精准调控和分析催化转化机理提供了依据[82]。
生物碳是一种通过将生物质热解产生的多孔碳质材料, 由于其高比表面积、 可调节的孔隙度、 化学惰性、 疏水性和丰富的碱性官能团而被认为是最有前途和可持续的CO2吸附材料之一[83]。 Li等通过ZnCl2+CO2改性从污水污泥和松木屑混合物中提取了纳米多孔生物碳, 并研究了其对CO2的捕集潜力。 采用原位FTIR漫反射技术分析了该混合生物碳对CO2的吸附状态, 发现在2 360~2 380 cm-1处有一个非常强烈的吸收峰, 这与物理吸附CO2的不对称拉伸有关, 并且强度随着吸附时间的增加而增加, 说明物理吸附在CO2捕获中起重要作用。 同时在1 551~1 744 cm-1处表现出弱峰值强度, 这归因于碳酸盐的形成, 表明化学吸附在CO2捕获过程中也起着积极的作用[84]。
生物质衍生分子因其来源广泛、 高容量和可持续性等特点, 已被确定为有机钾离子电池的可行阳极。 Qu等将天然核桃外果皮为原料制备的具有电化学活性的核桃酮与碳纳米管网络交联, 并进一步采用海藻酸钠作为关键粘结剂偶联并协同作用。 利用原位FTIR观察了放电-充电过程中C=O基团的演变, 揭示其电化学机理。 在放电过程中, 在1 630 cm-1处C=O基团的强度逐渐减弱, 而在1 050 cm-1处C— O基团信号逐渐增强, 因为K+离子可以与C=O基团配位形成C— O— K键, 从而降低了C=O键的比例, C— O键的数量增加。 在充电过程中, C=O和C— O基团的强度恢复到初始值, 表明C=O基团的还原和电极内钾的储存具有高可逆性[85]。
原位红外光谱技术在生物质热解、 化学催化和电催化转化等方面的研究开展提供了丰富的理论支撑。 通过实时监测反应过程中物质随反应条件的变化, 来跟踪反应体系结构和官能团的演变, 深入研究了反应路线和反应机制。 原位红外光谱技术有助于加深我们对生物质转化机理的理解, 为进一步设计更先进的转化途径提供有效指导。 然而, 进一步扩大原位红外光谱技术在生物质转化领域的应用仍然存在一些挑战和困难。 因此, 以下两方面是我们对表征生物质转化过程的原位红外光谱技术的展望:
(1)生物质通过精炼可以生成合成气、 液体燃料和高附加值化学品, 转化过程中会涉及大量的多相化学反应。 而目前原位透射和原位漫反射光谱技术主要应用于气-固催化反应的研究, 对于固-液等反应体系的研究存在许多限制, 原位衰减全反射红外光谱技术在高温高压等反应条件下易造成晶体损伤, 因此对原位反应池进行技术优化与突破, 提升极端条件适配性, 抑制水蒸气、 CO2等干扰, 有利于扩展其在生物质转化研究中的应用;
(2)另一方面是发展多模态联用技术以全面揭示反应机制。 将原位FTIR与质谱联用实时关联气相产物(如H2、 CO、 小分子烃类)与官能团演变, 并可以实现一些产物的定量分析。 将原位FTIR与拉曼光谱联用, 通过拉曼光谱检测非极性键弥补红外光谱盲区, 全面追踪碳骨架重构过程。 更进一步可以构建“ 红外-拉曼-质谱” 一体化反应池, 实现同一反应位点的化学键演变、 分子结构重构与产物逸出行为的同步解析。 多模态联用技术的深化发展将成为研究生物质转化的核心表征手段, 为实现碳中和目标提供关键技术支撑。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|