



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
非静水压环境对U3O8高压拉曼声子行为的影响
[王艺佳1  , 吴彬彬
, 吴彬彬1 , 刘静仪1 , 房雷鸣2 , 刘本琼2 , 雷力1, * ]
, 吴彬彬]
|
|


作者简介: 王艺佳, 女, 1999年生, 四川大学原子与分子物理研究所硕士研究生 e-mail: wyj18835734561@163.com
八氧化三铀(U3O8)是动力学和热力学最稳定的铀氧化物。研究U3O8的高压相稳定性和高压声子行为对其在核工业、 催化剂等领域的应用具有重要参考价值。由于铀原子([Rn]5f36d17s2)和氧原子([He]2s22p4)核外电子结构存在显著差异, 因此很难通过同步辐射X射线衍射技术探测高压下铀氧化合物中U—O 键合的细微变化。然而拉曼光谱对高压下U—O键合变化非常敏感, 它能揭示物质在高压下包括键合或者化学计量等方面的重要信息。目前对U3O8高压结构相变的研究通常聚焦于静水压环境, 而非静水压环境对U3O8高压相变的影响尚未得到深入研究。该研究基于金刚石对顶砧的高压拉曼散射技术探究了静水压和非静水压环境对正交α-U3O8高压相变与声子行为的影响。研究表明, 静水压(8.1 GPa)和非静水压(8.2 GPa)条件下α-U3O8高压相变起始压力很接近。但由于非静水压环境中样品内的微区偏应力较大, 因此非静水压环境中U3O8高压相变结束压力(16.4 GPa)比静水压环境中相应压力(18.5 GPa)约低2~3个GPa。同时本文给出了两对比实验中主拉曼模的一阶压力系数与Mode-Grüneisen参数 γ。对比分析可知, 高压相变前静水压条件下各主模零压下的一阶压力系数绝对值|d ω/d P|几乎均大于非静水压条件下的相应值, 这表明相变前非静水压环境中各拉曼模对压力响应不明显; 一旦高压相变开始, 由于较大微区偏应力的存在极大程度增强了铀氧原子外层电子的耦合作用, 故相变后非静水压下压力系数绝对值显著增大。零压下
The triuranium octoxide (U3O8) exhibits exceptional kinetic and thermodynamic stability among various uranium oxides. The study of the high-pressure phase stability and high-pressure phonon behaviour of U3O8 is an important reference for its application in the nuclear industry, catalysts, and other fields. Due to the complexity of the electron structure outside the nucleus of the uranium atoms ([Rn]5f36d17s2), compared to the oxygen atoms ([He]2s22p4), significant differences exist between their electron configurations. Therefore, synchrotron X-ray diffraction makes it difficult to detect subtle changes in U—O bonding for uranium oxides under high pressure. However, Raman spectroscopy is highly sensitive to changes in U—O bonding at high pressure, and it can reveal some important information about substances at high pressure, including bonding or stoichiometry. To date, the investigations on the high-pressure structural phase transition for U3O8 have typically focused on exploring its evolution in hydrostatic environments. However, a deep investigation of the effects of non-hydrostatic environments on the high-pressure phase transition of U3O8 has not been conducted yet. In this work, the effects of hydrostatic and non-hydrostatic environments on the high-pressure phase transition and phonon behaviour for orthorhombic α-U3O8 have been investigated using a high-pressure Raman scattering technique based on the diamond anvil cell. Our results show that initialization transition pressures of α-U3O8 under hydrostatic (8.1 GPa) and non-hydrostatic (8.2 GPa) conditions are very close. However, the significant micro zonation bias stress present within the sample in the non-hydrostatic environments leads to the completion transition pressure (16.4 GPa) being approximately 2 to 3 GPa lower compared to the corresponding values in the hydrostatic environments (18.5 GPa). The first-order pressure coefficients and mode-Grüneisen parameters γ of the main Raman modes in two comparison experiments were given. The results show that before the high-pressure phase transition, the absolute values of the zero-pressure first-order pressure coefficients |d ω/d P| for the main Raman modes under the hydrostatic environments are generally greater than those under the non-hydrostatic environments, which indicates that the Raman modes exhibit an insignificant response to the pressure under the non-hydrostatic environments. However, the absolute values of the first-order pressure coefficients under the non-hydrostatic environments are significantly larger once the high-pressure phase transition begins. This may be caused by significant micro zonation bias stress that greatly strengthens the mutual coupling between the outer electrons of the uranium-oxygen atoms. The
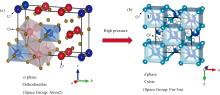
铀(U)是用于提供核裂变材料的重要元素之一。由于其原子结构中存在未被填满的5f电子壳层([Rn]5f36d17s2), 铀原子极易被氧化; 因此在核燃料循环过程中会形成多种不同的氧化物。在众多铀氧化物中, 八氧化三铀(U3O8), 在核工业中通常被称为“ 黄饼” [1], 是动力学和热力学最稳定的形式之一[2]。由于U3O8物化性质相对比较稳定, 在核工业、 催化剂等领域应用广泛[3, 4], 吸引了大量研究者对其结构[5, 6, 7]、 电子[8]、 热力学[9, 10, 11]和磁学[12]等特性进行研究。U3O8包含多种晶形, 其中在常温常压条件下的正交结构α 相(空间群Amm2)最为常见, 对其相关研究也最为广泛[6, 12, 13, 14, 15, 16, 17]。前期研究表明, 在高压下U3O8会发生由正交结构α 相向立方萤石型δ 相的结构转变(图1)。
 | 图1 U3O8的两种结构 (a): 空间群为Amm2的正交α 相; (b): 空间群为Fm-3m高压立方δ 相Fig.1 Schematic structure for U3O8 (a): The orthorhombic α -phase with space group Amm2; (b): The high-pressure cubic δ -phase with space group Fm-3m |
高压拉曼散射光谱和高压同步辐射X射线衍射技术是高压物质科学研究的两种重要实验手段。铀原子核外电子结构复杂, 且铀原子([Rn]5f36d17s2)和氧原子([He]2s22p4)核外电子结构存在显著差异; 因此, 很难通过同步辐射X射线衍射技术探测高压下铀氧化合物中U—O键合的细微变化。然而拉曼光谱对高压下U—O键合强度的变化非常敏感, 它可以揭示有关U—O的特殊价电子键合构型和化学计量等方面的重要信息。2011年, 美国Lawrence Livermore国家实验室的Lipp等借助高压拉曼散射技术探究了以氖为传压介质时U3O8的高压相变。他们发现, U3O8大约在7.7 GPa发生高压相转变, 并且室温加压和卸压过程中均无游离氧出现[5]。随后, 2014年美国密歇根大学的Zhang团队利用甲乙醇混合物作传压介质, 对α -U3O8进行了原位高压同步辐射XRD研究。他们的实验结果表明U3O8中具有萤石型结构的新高压相在高于8 GPa的压力下形成, 并且该高压相至少可稳定存在于40 GPa、 1 700 K的高温高压环境中[6]。2021年, 印度甘地原子研究中心的Balmukund Shukla等对不同晶粒尺寸的U3O8进行了原位高压和高压高温X射线衍射研究[7]。他们发现, 在静水压条件下对纳米和块体U3O8的加压会诱导U—O体系出现压致无序现象, 且相比于块体U3O8(相变起始压力6.4 GPa, 相变结束压力37.0 GPa), 纳米U3O8(相变起始压力5.2 GPa, 相变结束压力45.0 GPa)在高压相变过程中存在更广的压力共存区。此外, 一旦高压相变开始, 高温在增强高压相变动力学方面发挥着至关重要的作用[7]。与此同时, 计算物理的研究者们通过第一性原理计算, 也获得了不少关于U3O8高压行为的有趣结果。2011年, 中国工程物理研究院流体物理研究所的Geng等利用LSDA+U函数计算发现, 晶格振动会略微增加铀空位的数量, 而静水压则会改变所有缺陷的能量, 进而极大程度地改变缺陷数量[2]。2022年西北工业大学的Wang团队对α -U3O8的弹性和热力学性质进行了详细的第一性原理研究, 他们发现较于UO2和UO3, α -U3O8的弹性各向异性指数AU值最大(其中AU=0意味着晶体各向同性, 且AU值偏离0的程度越大, 晶体各向异性程度越高。), 表明α -U3O8的各向异性程度最高。同时弹性参数计算值表明c轴比a轴更易被压缩[18]。
高压相变是物质在高压条件下由一种状态转变为另一种状态的物理过程。通过研究相变过程中物质的热力学、 动力学等性质, 人们可以探究物质在高压下的结构和性质变化规律。相变研究不仅有助于探究物质的基础性质, 还在材料科学、 地球科学以及能源利用等领域发挥着重要作用[19, 20, 21, 22]。由压力引起的高压相变, 通常会改变常温常压下物质原有的物化性质。实际上高压相变的应用往往聚焦于非静水压环境。故探究静水压和非静水压环境对物质高压相变的影响, 为深入认识物质的高压演变行为提供了参考价值。
对于U3O8, 过去研究者们一般聚焦于探究静水压环境下该物质的结构相变, 而非静水压环境对U3O8高压相变的影响尚未得到深入研究。因此, 在本文中, 通过高压拉曼光谱对α -U3O8的高压行为进行了详细研究, 旨在探究非静水压环境对α -U3O8高压相变的影响。
本工作中使用的U3O8样品是通过加热铀制备得到, 购买自西安鼎天化工有限公司。我们首先使用粉末X射线衍射仪(PXRD, Haoyuan DX-2700 BH)对初始样品进行了XRD结构表征[图2(a)]。初始样品的X射线衍射峰与标准卡片中正交结构α -U3O8(空间群Amm2)的衍射谱吻合得很好。高压拉曼光谱实验在四川大学极端条件科学实验室(ESL)的高压拉曼光谱平台上完成。基于样品特性, 本工作选择532 nm固态激光器(RGB laser system NovaPro 300 mW)作为拉曼激发光源, 激光光斑尺寸大约5 μ m, 激光输出功率30 mW以下(样品表面入射的激光功率小于0.82 mW)。样品聚焦物镜的倍数为20X, 工作距离为30.5 mm, 通过光谱仪(Andor Shamrock SR-303i-B)和EMCCD(Andor Newton DU970P-UVB)采集不同压力下U3O8的拉曼光谱。拉曼实验前采用单晶硅片(520 cm-1)进行系统校正, 常温常压下α -U3O8的拉曼光谱如图2(b)所示。高压拉曼实验的典型采谱曝光时间为3 s, 积累次数为20次。
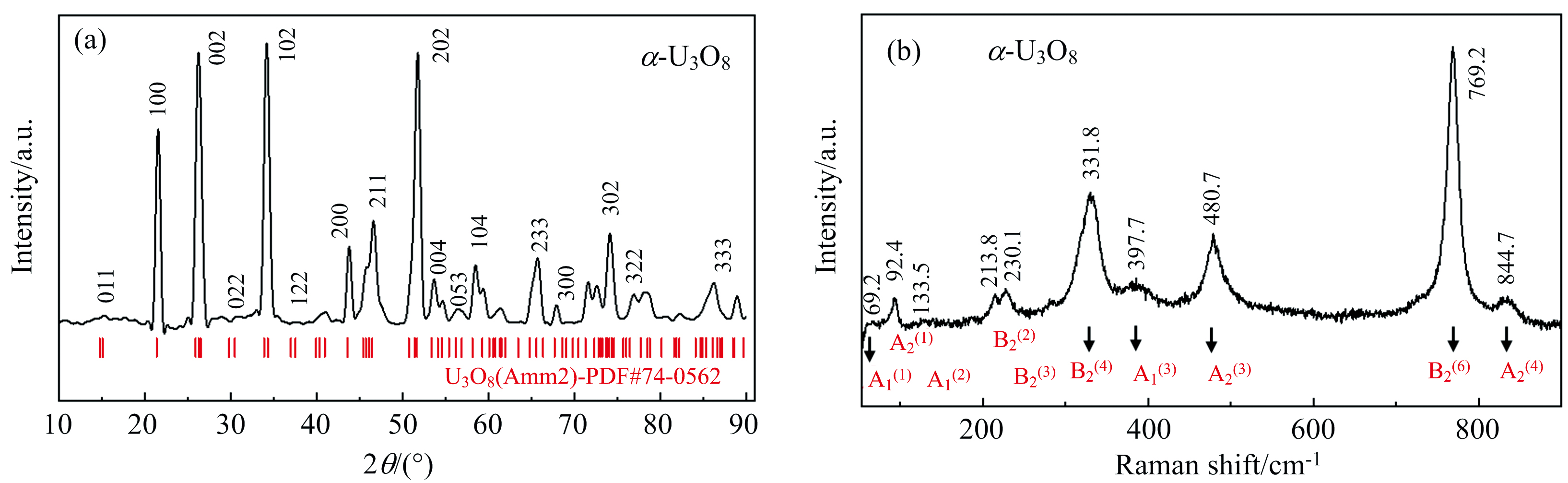 | 图2 在常温常压条件下α -U3O8的(a)XRD谱图和(b)拉曼光谱图Fig.2 (a) The XRD pattern and (b) Raman spectra of α -U3O8 at ambient conditions |
基于金刚石压砧(DAC)的高压拉曼散射光谱技术是高压拉曼谱学的主流实验技术[23, 24]。图3(a)是高压拉曼散射实验的实物图, 图3(b)是高压拉曼实验中DAC样品组装示意图, 初始U3O8样品呈黄绿色[图3(c)]。为了减弱背底和散射效应, 本工作采用砧面直径为 500 μ m的IIA高纯度金刚石对顶砧来提供高压环境, T301不锈钢压砧封垫的预压厚度约为30~40 μ m, 激光打孔的样品腔直径约为120~150 μ m。静水压实验采用体积比为4∶ 1的甲乙醇混合物作为传压介质, 而非静水压实验不采用任何传压介质。高压实验的压力通过红宝石R1荧光峰随压力的偏移进行标定[25]。实验中采集的所有拉曼光谱数据均通过PeakFit软件进行拟合处理。
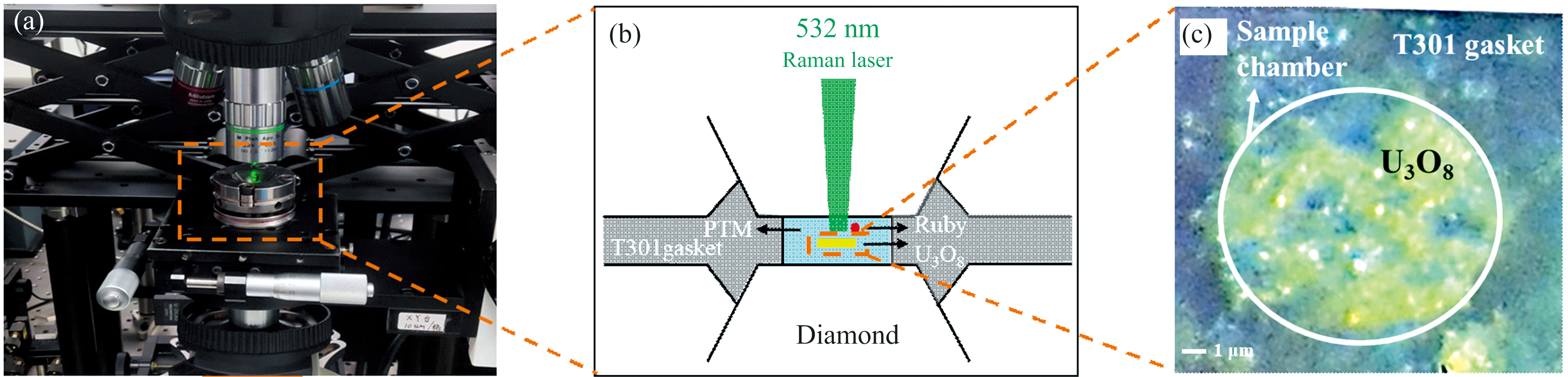 | 图3 (a) 高压拉曼光谱实验样品台; (b) DAC样品组装示意图; (c) DAC样品腔的光学照片Fig.3 (a) High-pressure Raman spectroscopy experimental sample stage; (b) the DAC sample assembly schematic; (c) the optical photograph of the DAC sample chamber |
图2(b)显示了常温常压条件下α -U3O8的拉曼谱图。C2v点群分析表明, 空间群为Amm2的α -U3O8应该具有33个拉曼激活模, 其不可约表示为Γ =11A1+4A2+7B1+11B2。通过参考Butler、 郭琳媛等[26, 27]的U3O8拉曼振动模数据, 我们对拉曼光谱中观测到的10个拉曼峰进行了振动模认定[图2(b)], 特别选择了拉曼峰较强且分辨率较高的5个拉曼模(
为提供较好的静水压环境, 我们选择体积比为4∶ 1的甲乙醇混合物(~10 GPa固化)作DAC传压介质。在室温下, 把初始α -U3O8加压到18.5 GPa, 直到样品无明显拉曼信号[图4(a)]。空间群为Fm-3m的立方高压相并不存在一阶拉曼振动模。为了比较静水压和非静水压环境对α -U3O8高压相变的影响, 我们再次进行了无传压介质时的高压拉曼散射实验, 并把压力加载到18 GPa以上[图4(b)]。通过对比两次实验的拉曼光谱数据, 发现各拉曼模随着压力增加均出现拉曼峰强的弱化和峰型的宽化。此外, 可以清晰地看到, 两实验中低压下
 | 图4 (a) 静水压环境下α -U3O8的高压拉曼光谱; (b) 非静水压环境下α -U3O8的高压拉曼光谱 ↑ : 加压过程; ↓ : 卸压过程Fig.4 (a) Raman spectra of α -U3O8 under hydrostatic pressure condition and (b) under non-hydrostatic pressure condition ↑ : The compression process; ↓ : The decompression process |
为了更直观地确定非静水压条件对α -U3O8高压相变的影响, 图5给出了两次比较实验中
 | 图5 静水压(粉色)与非静水压(蓝色)高压实验中α -U3O8四个主拉曼模( 四张插图分别代表四个主拉曼模的原子位移; 子图中两条灰色虚线分别代表高压相变中相变起始压力(左)和相变结束压力(右); 灰色矩形代表高压相变过程中由初始正交相向高压立方相转变的压力共存区Fig.5 Comparison of the pressure dependence of the four Raman modes during compression with hydrostatic (pink) and non-hydrostatic (blue) pressure conditions for α -U3O8: The four insets represent the atomic displacements of four main Raman modes, respectively; The two grey dotted lines in the subfigures represent initialization (left) and completion (right) transition pressures for the high-pressure phase transition, respectively; The grey rectangular represents the pressure coexistence regime from the initial orthorhombic structure to the high-pressure cubic structure during the high-pressure phase transition |
同时, 当样品处于非静水压条件时, 高压相变起始压力约为8.2 GPa。可以注意到, 不同于其他拉曼模随压力变化的频移方向,
但是, 从图4和图5中可以看出, 非静水压常温高压实验中各主模存在的最高压力Pf小于静水压常温高压实验中相应值。具体来说, 非静水压环境下, 当外界施加压力超过16 GPa时, 高压拉曼谱图中除了低波数处单峰外, 其余拉曼峰消失; 而静水压环境下当压力高达18 GPa以上时, 高压拉曼谱中所有拉曼峰消失。这表明, 非静水压环境中U3O8相变结束压力比静水压环境中相应压力约低2~3个GPa。由于无传压介质确保腔内样品受到的压力各向相同, 故非静水压环境中样品内的微区偏应力较大, 这可能是非静水压下U3O8高压相变结束压力较低的主要原因。此外, 图5中灰色矩形区域代表高压相变时由初始正交结构向高压立方结构转变的压力共存区, 这表明该高压相变过程较缓慢。
相比于其他研究者借助高压同步辐射技术确定的静水压下U3O8高压相变结束压力[6, 7], 本工作通过高压拉曼技术确定的相变结束压力值略低(18.5 GPa)。拉曼散射技术中通常使用的激光光斑尺寸仅有几个μ m, 而同步辐射实验中聚焦后的光斑尺寸在几十μ m以上。由于同步辐射光源光斑尺寸较大, 造成实验过程中探测到的样品尺寸范围较广, 因此相比于激光微区拉曼散射技术, DAC同步辐射实验中存在更大的压力梯度, 这可能导致研究者们在确定U3O8高压相变结束压力时与本工作的实验结果相比存在滞后。
在表1中, 我们比较了两次实验中
| 表1 在静水压和非静水压室温加压过程中零压下α -U3O8拉曼峰位置ω 0(Raman shift/cm-1)、 一阶压力系数(dω /dP)P=0以及Mode-Grü neisen参数γ 0 Table 1 Zero-pressure Raman peak position ω 0 (Raman shift/cm-1), the first-order pressure coefficient (dω /dP)P=0, and mode-Grü neisen parameter γ 0of α -U3O8 during compression at room temperature with hydrostatic and non-hydrostatic pressure conditions |
在实验误差范围内, 拉曼波数ω 的压力依赖性可以用下述公式很好地描述: ω (P)=ω 0+aP+bP2。其中, 拉曼波数ω 的单位是cm-1, 压力P的单位是GPa, ω 0、 a、 b分别表示零压力下的拉曼波数、 一阶压力系数和二阶压力系数。系数a通常用(dω /dP)P=0的形式表示, 这有助于确定零压下的Mode-Grü neisen参数
式(1)中, Vm和B0分别为零压力下的摩尔体积和体弹模量。如表1所示, 本文给出了各拉曼主模零压下的拉曼波数ω 0, 一阶压力系数(dω /dP)P=0和Mode-Grü neisen参数γ 0。
Mode-Grü neisen参数反映固体的非谐性, 并决定着物质的热膨胀, 它可以通过声子能量和晶胞体积间的关系来计算。Mode-Grü neisen参数还能反映温度对晶胞体积或晶格动力学方面的影响[23, 24, 28]。数值较大的Mode-Grü neisen参数反映了较强的原子间非谐相互作用[29], 而趋于0的Mode-Grü neisen参数则意味着晶格振动趋于严格的简谐。本工作中, 伴随压力增加, γ 为正的
此外, 借助Zhang等的高压X射线衍射实验数据[6]和Miskowiec等的高温X射线实验数据[11], 我们进一步研究和分析了U3O8在高压下的可压缩性以及在高温下对温度的响应行为。基于Zhang等报道的α -U3O8在不同压力下的晶胞参数值, 可以注意到, 高压相变前, 沿b轴和c轴的晶格轴比(b/b0和c/c0)随着压力增加呈现相似的衰减趋势。然而, 沿a轴的晶格轴比(a/a0)随压力增加而衰减的斜率逐渐增大。因此, 沿a、 b和c轴的压缩行为表明α -U3O8低压正交相具有各向异性。同时, 沿不同晶轴方向的结构观测结果显示[如图1(a)], 相同多面体沿a轴首尾顺次相连, 而以U1和U2原子为中心的多面体则沿b轴和c轴交错分布。这表明相较于b轴和c轴, a轴不易被压缩。另一方面, 在室温加压时
基于金刚石对顶砧静高压技术, 分别在静水压和非静水压条件下对α -U3O8的拉曼振动模对高压的响应行为和相稳定性开展了对比研究。以拉曼模频移随压力的不连续变化为典型特征, 研究发现: (1)静水压和非静水压条件下α -U3O8高压相变起始压力很接近; (2)但由于非静水压环境中样品内的微区偏应力较大, 故非静水压环境中U3O8高压相变结束压力低于静水压环境中对应压力; (3)高压相变前, 与静水压环境相比, 非静水压环境中各拉曼模对压力响应不明显; 一旦相变开始, 非静水压环境中较大微区偏应力的存在, 极大程度地增强了铀氧原子外层电子间的相互耦合作用, 这使得相变后非静水压环境中拉曼模一阶压力系数绝对值显著增强。(4)各拉曼模压力系数绝对值的对比结果表明高压相变前α -U3O8晶胞内, a轴比b、 c轴更难被压缩。本工作首次深入探究了静水压和非静水压环境对α -U3O8高压相变的影响, 对了解α -U3O8高压相稳定性和晶格动力学行为具有重要参考价值。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|