







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
萃取分光光度法测定氯化钾体系中的十八胺
[宋忠梅1, 4  , 路淼
, 路淼1, 4 , 张慧芳1, * , 张鹏瑞1 , 房得珍1, 4 , 马亮1, 4 , 刘青青2 , 于雪峰2 , 刘海宁1 , 叶秀深1 , 马珍2 , 彭文博3 , 吴志坚1 ]
, 路淼, 张鹏瑞|
|


作者简介: 宋忠梅, 女, 1999年生, 中国科学院青海盐湖研究所硕士研究生 e-mail: songzhongmei21@mails.ucas.ac.cn
十八胺是冷结晶-正浮选法生产氯化钾的常用捕收剂, 用量显著影响浮选分离效率。 由于其会吸附在氯化钾表面, 不可避免地在氯化钾中残留, 不利于高纯钾盐产品的开发。 针对氯化钾生产过程及产品中十八胺含量测定的需求, 依据十八胺和溴酚蓝通过范德华力和氢键络合的作用原理, 开发了以乙酸丁酯为萃取剂、 溴酚蓝钠盐为显色剂的萃取分光光度法测定氯化钾中十八胺的浓度。 考察了溶液pH、 十二烷基吗啉、 共存盐、 平衡时间、 显色剂用量对十八胺浓度测量的影响。 结果表明: 当溶液pH从3~9逐渐增大时, 十八胺与溴酚蓝的络合作用减弱且络合分子极性增大, 导致吸光度降低; 在pH<5时, 十二烷基吗啉和溴酚蓝能形成有色络合物, 当pH为6~9时萃取液吸光度值趋近于零; 共存氯化钾、 氯化钠、 硫酸钾、 氯化镁等溶液的离子强度增大, 氢键作用减弱但络合分子的盐析效应增强, 溶液中微量的Li+、 NH4+和B对萃取液吸光度影响不大; 显色剂用量过大时, 吸光度值太大且不符合朗伯-比尔定律, 平衡时间影响较小。 选择测定条件为: 溶液的pH为6、 离子强度为1 mol·L-1, 2 mmol·L-1显色剂的用量为0.50 mL, 采用缓冲溶液定容至25 mL, 反应5 min后加入5.00 mL乙酸丁酯萃取, 待分层平衡2 min后于458 nm处测定萃取液吸光度, 工作曲线为 A=0.049 49 c+0.066 24( R2=0.992 3, ε=1.33×104 L·mol-1·cm-1, 0~10 mg·L-1)。 该方法的相对标准偏差为0.33%~2.63%, 平均相对误差为-0.90%, 十八胺和十二烷基吗啉混合体系的平均相对误差为-0.25%。 采用该方法测得正浮选精钾母液中十八胺含量为8.66 mg·L-1, 加标回收率为95.5%~106%, 可用于测定盐湖钾肥生产过程中十八胺的浓度。
, LU Miao, ZHANG Peng-ruiOctadecylamine is a flotation collector commonly used to produce potassium chloride by cold crystallization-positive flotation, and its dosage will significantly affect the flotation separation efficiency. In addition, octadecylamine will adsorb on the surface of potassium chloride and inevitably remain in potassium chloride products, which is inconducive to developing high-purity potassium salt products. To meet the requirement of determination of octadecamide potency in potassium chloride, an extraction spectrophotometry was developed, based on the principle of van der Waals force and hydrogen bonding between octadecylamine and bromophenol blue with butyl acetate as extractant and bromophenol blue sodium salt as chromogenic agent. The effects of solution pH, dodecyl morpholine, co-existing salt, equilibrium time, and the amount of chromogenic agent on the concentration measurement of octachylamine were investigated. The results show that when the solution pH increases from 3 to 9, the absorbance of the extract decreases because the complexation between octachylamine and bromophenol blue weakens. The polarity of the complex molecules increases. When the solution pH is less than 5, dodecyl morpholine and bromophenol blue can also form colored complexes. While pH is between 6 and 9, the absorbance tends to zero. The ionic strength of the coexisting solutions of potassium chloride, sodium chloride, potassium sulfate and magnesium chloride increases, weakening the hydrogen bonding. Still, the salting-out effect of the complex molecules is enhanced, and the trace Li+, NH4+ and B in the solution have little effect on the absorbance. The absorbance does not conform to Lambert-Beer law when the amount of chromogenic agent is too large. Besides, equilibrium time has little effect on the absorbance. The determination conditions in potassium chloride solution are as follows: The solution pH is 6, the ionic strength is 1 mol·L-1, and the amount of 2 mmol·L-1 chromogenic agent is 0.5 mL. 5 mL butyl acetate is added to the 25 mL aqueous solution adjusted by buffer after 5 min of reaction for extraction. After 2 min of delamination equilibrium, the absorbance of the extraction solution is tested at 458 nm. The working curve is A=0.049 49 c+0.066 24 ( R2=0.992 3, ε=1.33×104 L·mol-1·cm-1, 0~10 mg·L-1). The relative standard deviation of this method was 0.33%~2.63%, the mean relative error was -0.90%, and the mean relative error of the system of octadecylamine and dodecylmorpholine was -0.25%. The content of octadecamide in the filter liquor derived from washing potassium chloride after positive flotation was 8.66 mg·L-1 and recoveres were 95.5%~106% by this method. The extraction spectrophotometry has been proven to be appropriate for detecting the concentration of octadecamide in the production of potassic fertilizer in salt lakes.
青海察尔汗盐湖是我国重要的钾肥生产基地, 氯化钾产能达到800万吨/年, 为保障国家粮食安全做出了重要贡献。 目前氯化钾生产工艺主要有冷分解-正浮选法和反浮选-冷结晶法。 十八胺(ODA)是常用的正浮选捕收剂, 用量会显著影响浮选分离效率, 由于其会吸附在氯化钾表面, 也会不可避免的在氯化钾产品中残留, 不利于高纯钾盐产品的开发[1, 2]。 因此, 建立一种便捷可靠的测定氯化钾中十八胺的分析方法是很有必要的。
目前针对十八胺的分析方法主要有气相色谱法[3, 4, 5]和分光光度法[6, 7, 8, 9, 10], 其中分光光度法因其操作简便、 分析成本低在分析检测中得到广泛的应用。 刁祥瑞、 梁慧斌等[6, 10]采用氯仿为萃取剂, 溴甲酚绿、 金橙-2分别作为显色剂测定了氯化钾溶液中十八胺的含量。 氯仿具有一定毒性极易挥发, 长期使用对操作人员有健康危害, 且受危险化学品和易制毒化学品的管控, 购买、 使用和存放均有严格要求, 导致此类方法的应用受到限制。 李兵、 葛红花等[7, 9]用毒性较低的乙酸乙酯、 乙酸丙酯代替氯仿, 采用甲基橙为显色剂测定了水中十八胺的含量。 由于实际氯化钾浮选分离体系中通常有镁离子, 而甲基橙与镁离子发生络合沉淀反应, 干扰测定。 胡家元等[8]提供了一种不添加显色剂的十八胺浓度测定方法, 在待测溶液中加入冰乙酸, 直接测定紫外区吸光度, 对10~50 mg· L-1含量范围的十八胺能有效测定。 十八胺在水中溶解度较小, 由于盐析效应, 十八胺在氯化钾溶液中溶解度更小, 该方法并不适用于氯化钾中十八胺含量的测定。 前期提出采用烷烃为萃取剂、 溴酚蓝钠盐为显色剂测定氯化钾中十八胺含量的方法, 可避免镁离子与显色剂的络合沉淀且萃取剂毒性低[11]。 由于多年来察尔汗盐湖矿区正浮选和反浮选工艺并行, 矿区内排放的尾矿中通常是正浮选捕收剂十八胺、 反浮选捕收剂十二烷基吗啉(DMP)共存, 十二烷基吗啉的存在会影响十八胺的测定。
对比研究现有方法的萃取体系(补充材料), 确定了以溴酚蓝钠盐为显色剂、 乙酸丁酯为萃取剂, 通过调控溶液pH避免十二烷基吗啉对十八胺测定的影响, 建立了氯化钾体系中十八胺含量的测定方法。
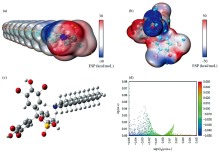
脂肪胺与阴离子染料形成可被有机溶剂萃取的有色络合物[12, 13]。 使用Multiwfn软件对十八胺和溴酚蓝的表面静电势进行了分析, 如图1(a)、 (b)所示十八胺和溴酚蓝表面静电势的分布情况, 红色区域表示静电电势为正, 蓝色区域表示静电电势为负。 对十八胺和溴酚蓝分子的络合进行了模拟, 如图1(c)、 (d)所示。 通过对十八胺分子和溴酚蓝分子作用进行独立梯度模型[14]模拟计算显示, 十八胺与溴酚蓝分子可通过分子间范德华力作用和氢键作用形成络合物。 如图1(d)所示δ g与sign(λ 2)p(r)的散点图反映了相互作用的强弱和类型, 在sign(λ 2)p(r)为0附近出现的峰值表示十八胺和溴酚蓝可通过范德华力相互作用络合, 以及左侧少量蓝色区域表示可通过氢键作用络合。
 | 图1 十八胺-溴酚蓝络合分子的化学计算分析 (a): 分子表面静电势分布; (b): 溴酚蓝分子表面静电势分布; (c): 十八胺-溴酚蓝络合物分子模拟; (d): 十八胺-溴酚蓝络合分子间相互作用IGM等值面图Fig.1 Chemical calculation and analysis of octadecamine-bromophenol blue complex molecule (a): Molecular electrostatic potential of octadecylamine; (b): Molecular electrostatic potential of bromophenol blue; (c): Molecular simulation diagram of octadecamine-bromophenol blue complex; (d): IGM isosurface diagram of intermolecular interaction between octadecylamine bromophenol blue complex molecules |
采用TU-1810PC型紫外-可见分光光度计和ME204型梅特勒电子天平。 试剂包括十八胺、 溴酚兰钠盐、 乙酸丁酯、 磷酸二氢钠、 氢氧化钠、 无水乙酸钠、 冰乙酸、 氯化钾、 氯化钠、 氯化镁、 硫酸钾、 氯化锂、 四硼酸钠、 氯化铵, 均为分析纯。 十二烷基吗啉为工业级、 正浮选精钾母液为青海盐湖工业股份有限公司提供, 实验用去离子水。
1.3.1 十八胺储备液
称取一定质量的十八胺并加入一定去离子水在80 ℃下水浴溶解, 加入盐酸酸化后用去离子水配制成50 mg· L-1的储备液, 使用时再配制成不同浓度的工作液。
1.3.2 十二烷基吗啉储备液
称取一定质量的十二烷基吗啉并加入去离子水, 加入盐酸酸化用去离子水配制成50 mg· L-1的储备液, 使用时再配制成不同浓度的工作液。
1.3.3 缓冲溶液
使用0.1 mol· L-1醋酸溶液和0.2 mol· L-1醋酸钠溶液依次配制出pH为3、 4、 5的醋酸-醋酸钠缓冲液; 使用0.4 mol· L-1磷酸二氢钠溶液和0.4 mol· L-1氢氧化钠溶液依次配制出pH为6、 7、 8、 9的磷酸二氢钠-氢氧化钠缓冲液。
1.3.4 显色剂溶液
称取0.142 8 g溴酚蓝钠盐溶于去离子水中, 于100 mL容量瓶中定容, 配制成2 mmol· L-1溶液。
1.4.1 溶液pH对十八胺、 十二烷基吗啉萃取体系吸收光谱的影响
分别配制pH为3、 4、 5、 6、 7、 8、 9三组浓度(12.5、 25.0、 37.5 mg· L-1)的十八胺和十二烷基吗啉溶液。 各取10 mL不同浓度的十八胺或十二烷基吗啉溶液于25 mL容量瓶, 加入0.5 mL 2 mmol· L-1显色剂溶液, 并用pH为3~9的缓冲溶液定容。 反应5 min后加入5 mL乙酸丁酯萃取有色络合物, 静置待分层后, 取适量萃取液于石英比色皿, 在190~800 nm波长范围进行光谱扫描。
1.4.2 盐对十八胺萃取体系吸收光谱的影响
取10 mL pH为6的十八胺溶液于25 mL容量瓶, 加入0.5 mL 2 mmol· L-1显色剂溶液, 分别准确称取不同质量的氯化钾、 氯化钠、 氯化镁、 硫酸钾、 氯化锂、 氯化铵、 四硼酸钠, 用pH为6的缓冲溶液溶解并定容。 反应5 min后加入5 mL乙酸丁酯萃取有色络合物, 静置待分层后, 取适量萃取液于石英比色皿, 在190~800 nm波长范围进行光谱扫描。
1.4.3 平衡时间对十八胺萃取体系吸收光谱的影响
取10 mL pH为6的十八胺溶液于25 mL容量瓶, 加入0.5 mL 2 mmol· L-1显色剂溶液, 加入氯化钾使离子强度为1 mol· L-1, 用pH为6的缓冲溶液溶解并定容。 反应5 min后加入5 mL乙酸丁酯萃取有色络合物, 静置待分层后计为起始时间, 继续静置0、 2、 5、 10、 15、 30和60 min, 取适量萃取液于石英比色皿, 在190~800 nm波长范围进行光谱扫描。
1.4.4 显色剂用量对十八胺萃取体系吸收光谱的影响
取10 mL pH为6的十八胺溶液于25 mL容量瓶, 分别加入0、 0.10、 0.25、 0.50、 0.75、 1.00、 1.50和2.00 mL 2 mmol· L-1显色剂溶液, 加入氯化钾使离子强度为1 mol· L-1, 用pH为6的缓冲溶液溶解并定容。 反应5 min后加入5 mL乙酸丁酯萃取有色络合物, 静置待分层后平衡2 min, 取适量萃取液于石英比色皿, 在190~800 nm波长范围进行光谱扫描, 并以去离子水做空白对照。
1.4.5 十八胺萃取体系的最佳吸收波长确定
取10 mL pH为6的不同浓度十八胺溶液于25 mL容量瓶, 加入0.5 mL 2 mmol· L-1显色剂溶液, 加入氯化钾使离子强度为1 mol· L-1, 用pH为6的缓冲溶液溶解并定容。 反应5 min后加入5 mL乙酸丁酯萃取有色络合物, 静置待分层后平衡2 min, 取适量萃取液于石英比色皿, 在190~800 nm波长范围进行光谱扫描。
2.1.1 溶液pH对十八胺萃取体系吸收光谱的影响
溶液pH对十八胺萃取体系吸收光谱的影响如图2(a, b, c)所示。 三组浓度的光谱变化趋势随着pH的变化基本一致。 在pH为3~5时吸光度具有较高的响应, pH大于5吸光度值降低, 到pH为8、 9的吸光度值整体趋近于零。 溴酚蓝钠盐的变色范围在3~4.6, 根据酸碱指示剂变色原理可知, 当pH小于5时, 溴酚蓝钠盐可转变成中性分子结构的溴酚蓝, 能够与十八胺分子通过氢键作用和范德华力进行络合。 图中光谱波长范围小于波谷490 nm可以看出, 十八胺和溴酚蓝更易通过范德华力作用而被萃入有机相。 而当光谱大于490 nm波长范围时, 氢键作用产生了此处的波峰, 络合物氢键的强弱与氢键的给予体和接受体有关, 溴酚蓝分子上C原子电负性较小, 与十八胺形成的氢键作用弱(C— H…N), 易受测定环境如温度等影响; 当十八胺的浓度较低时, 与溴酚蓝钠盐形成氢键作用的十八胺较少, 氢键作用力较弱不稳定, 出现了不规律的情况。 随着浓度增大至10和15 mg· L-1时, 能够与溴酚蓝钠盐形成氢键的十八胺增加, 光谱变化在490~650 nm波长范围内趋势一致, 呈现减弱趋势。 这是因为在酸性条件下, 氢离子的增加使十八胺被质子化, 从而削弱了十八胺和溴酚蓝分子的氢键作用, 溶液pH从3增加至5, 对氢键作用削弱的能力逐渐减弱, 490~650 nm波长范围的光谱逐渐增强。 当pH大于5时, 十八胺络合物光谱响应整体逐渐降低, 一方面, 溴酚蓝分子氢的电离程度增加, 导致络合分子极性增大, 使得十八胺络合物不易萃入有机相; 此外, 与十八胺氢键作用的溴酚蓝中性分子逐渐减少, 使得十八胺络合物分子减少, 从而导致吸光度值降低。
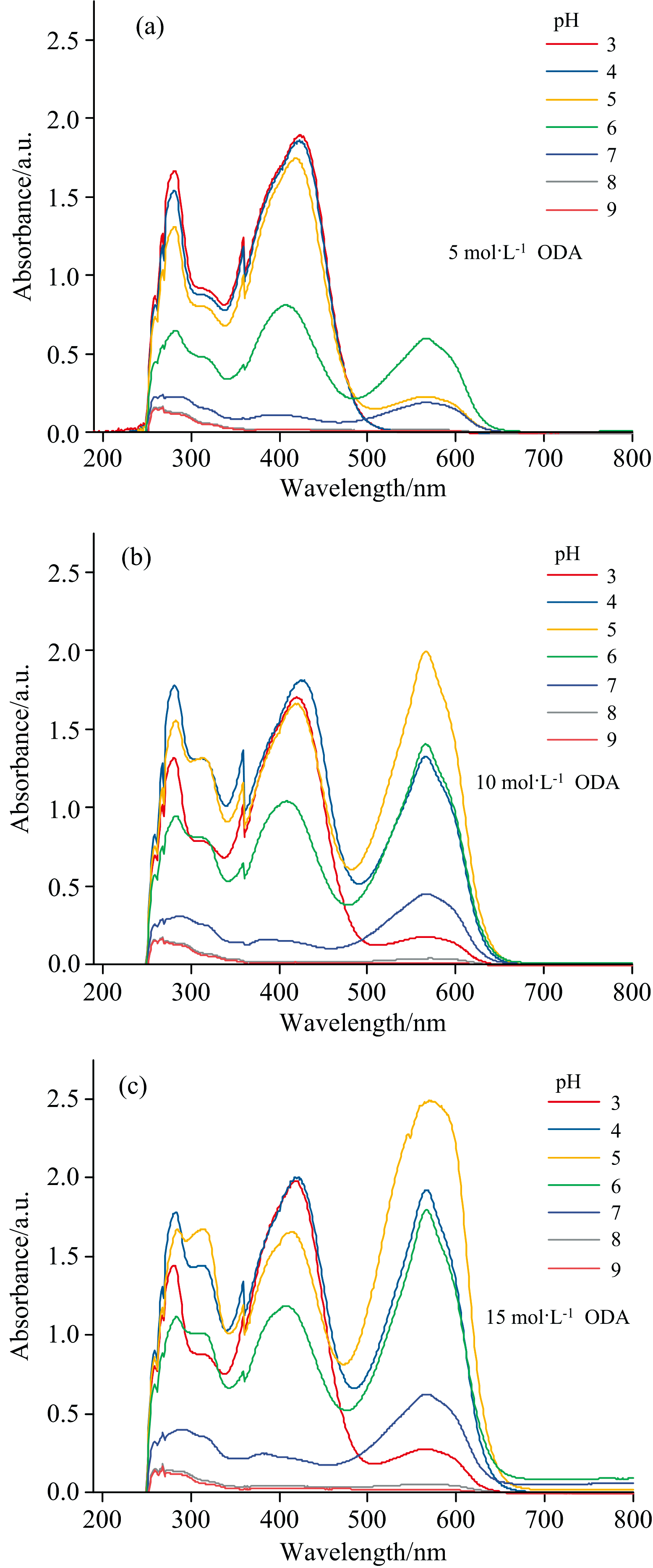 | 图2 溶液pH对十八胺萃取体系吸收光谱的影响 (a): 5 mol· L-1; (b): 10 mol· L-1; (c): 15 mol· L-1Fig.2 Effect of solution pH on absorption spectrum of octadecylamine extraction system (a): 5 mol· L-1; (b): 10 mol· L-1; (c): 15 mol· L-1 |
2.1.2 溶液pH对十二烷基吗啉萃取体系吸收光谱的影响
溶液pH对十二烷基吗啉萃取体系吸收光谱的影响如图3(a, b, c)所示。 三组浓度的光谱变化趋势基本一致, 吸光度值随pH增大而减小。 与十八胺相比, 十二烷基吗啉在pH为3~4时吸光度具有较高的响应, pH大于4吸光度值降低, 到pH为6~9的吸光度值整体趋近于零。 对比图2, 在溶液pH为6时十八胺络合物具有较好的光响应。 因此实验中选择测定pH为6, 可避免十二烷基吗啉对十八胺含量测定的影响。
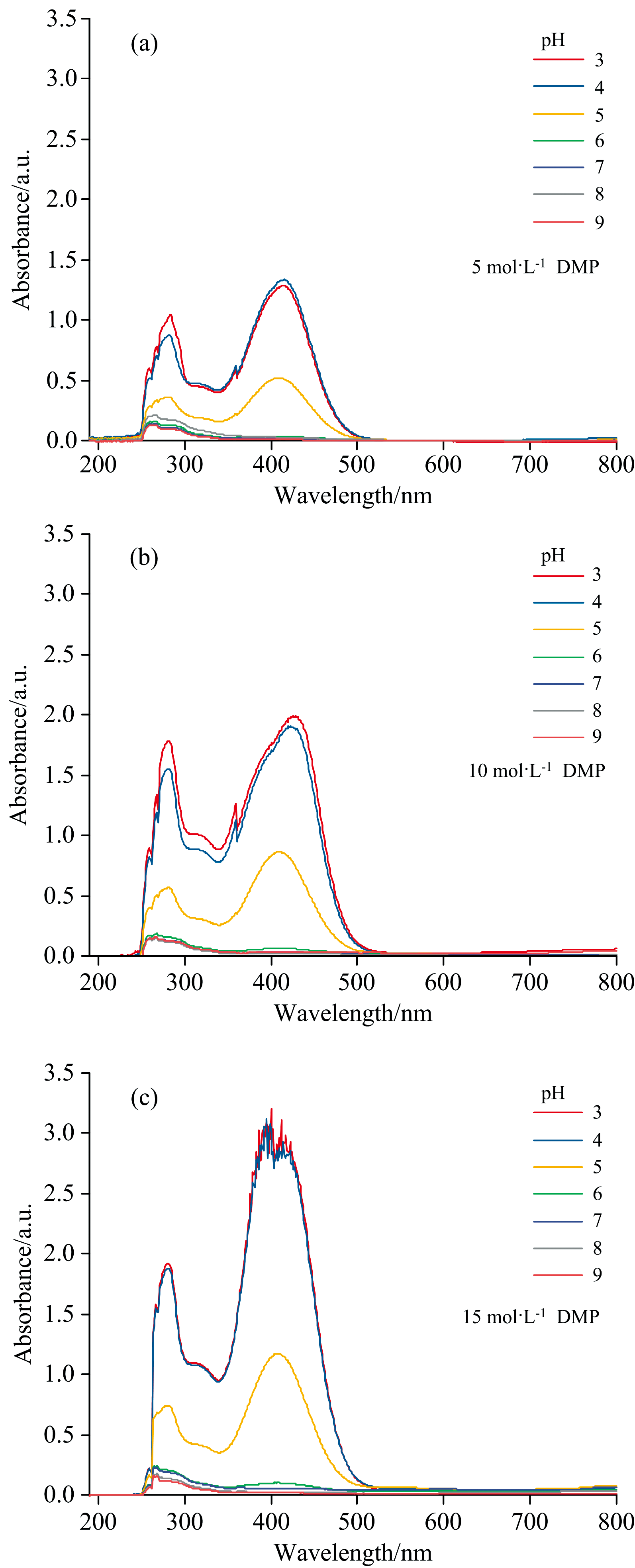 | 图3 溶液pH对十二烷基吗啉萃取体系吸收光谱的影响 (a): 5 mol· L-1; (b): 10 mol· L-1; (c): 15 mol· L-1Fig.3 Effect of solution pH on absorption spectrum of dodecylmorpholine extraction system (a): 5 mol· L-1; (b): 10 mol· L-1; (c): 15 mol· L-1 |
在钾肥生产过程中, 通常存在K+、 Na+、 Mg2+、 Cl-、 SO42-等离子, 以及少量的Li+、 硼(以B元素计)。 另外在生产实践中也发现存在少量NH4+。 因此分别考察了不同离子强度(
2.2.1 不同离子强度盐溶液对十八胺萃取体系吸收光谱的影响
不同离子强度盐溶液对十八胺萃取体系吸收光谱的影响如图4(a— f)所示。 溶液离子强度增加, 十八胺络合物在小于490 nm范围内吸光度增大, 由于盐析作用, 可以促进十八胺络合物萃入有机相。 在氯化钾、 氯化钠、 氯化镁溶液中, 光谱在大于490 nm范围波长呈现减弱趋势, 而硫酸钾溶液中光谱在大于490 nm范围几乎不变。 由于Cl-电荷密度比SO42-电荷密度小[15], 根据软硬酸碱理论, Cl-比SO42-更容易与溴酚蓝分子相互作用, 从而削弱了溴酚蓝分子与十八胺的氢键作用, 改变了十八胺络合物的络合方式。 如图4(e)所示, 不同离子强度盐溶液的光谱曲线在250~500 nm处部分重合, 说明离子强度在大于0.5 mol· L-1后继续增大对重合部分光谱影响较小。 选择离子强度为1 mol· L-1, 对不同盐溶液中十八胺络合物光谱的不同波长下进行吸光度相对标准偏差计算, 如图4(f)所示波段一329~381 nm和波段二453~470 nm的相对标准偏差小于5%, 表明不同盐在离子强度为1 mol· L-1条件下对这两段波长范围的吸光度值影响较小。 鉴于以上分析, 本研究选择离子强度1 mol· L-1的氯化钾当量。
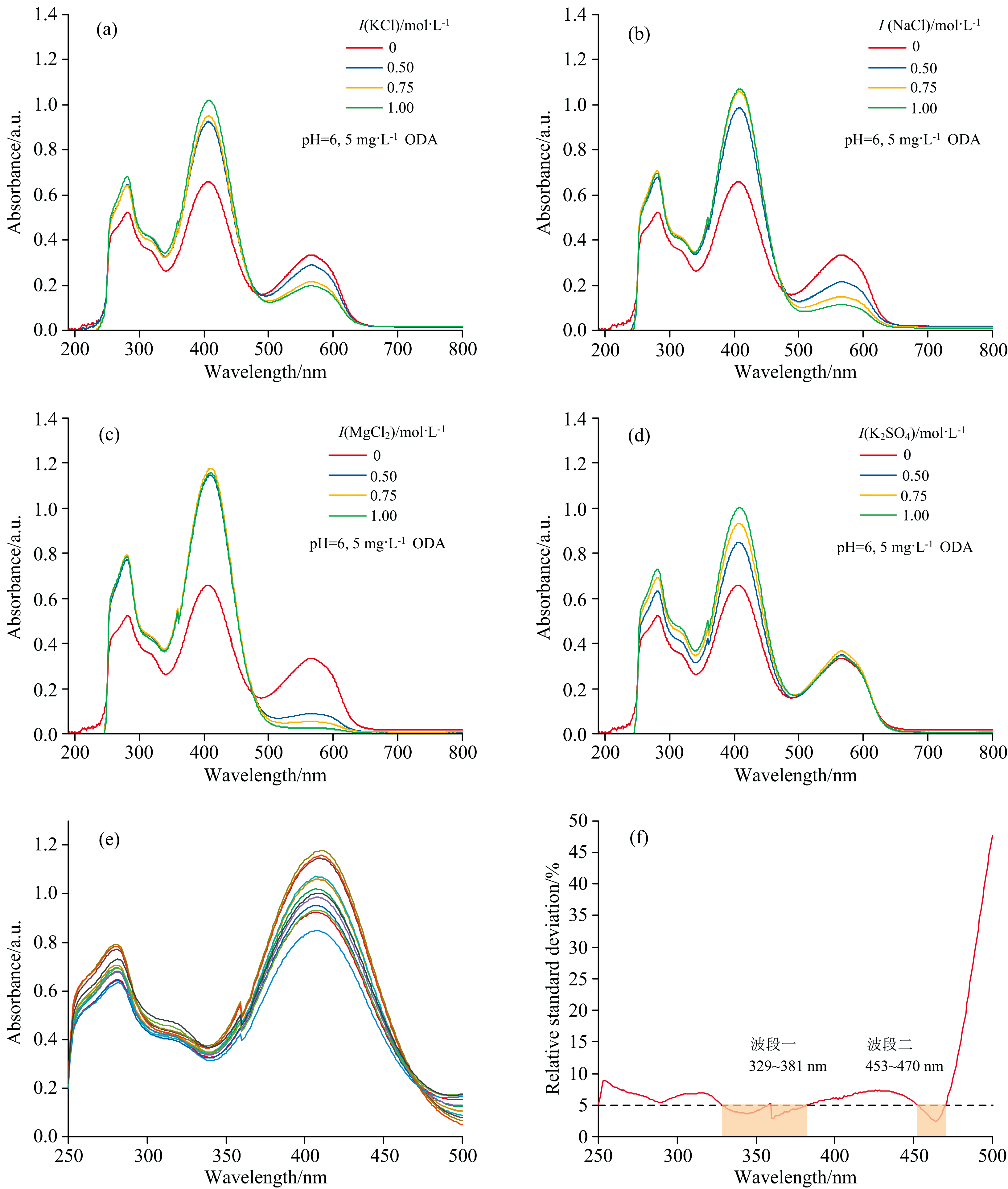 | 图4 不同离子强度盐溶液对十八胺萃取体系吸收光谱的影响 (a): KCl溶液; (b): NaCl溶液; (c): MgCl2溶液; (d): K2SO4溶液; (e): 不同离子强度盐溶液光谱; (f): 不同离子强度盐溶液光谱相对偏差Fig.4 Effect of salt solutions with different ionic strengths on the absorption spectrum of octadecylamine extraction system (a): Potassium chloride solution; (b): Sodium chloride solution; (c): Magnesium chloride solution; (d): Potassium solution; (e): Spectral curves of different ion stength salt solution at 250~500 nm; (f): Relative standard deviation of spectral curves of different ion strength salt solutions at 250~500 nm |
2.2.2 盐湖微量组分对十八胺萃取体系吸收光谱的影响
不同浓度微量组分对十八胺萃取体系吸收光谱的影响如图5(a— d)所示。 随着Li+和NH4+浓度的升高, 微弱的盐析作用促进光谱曲线响应增强但幅度较小。 除不加盐的基线外, 三组浓度的光谱曲线基本重合, 在波段一和波段二范围内的吸光度值波动较小, 说明微量Li+和NH4+对十八胺络合物光谱的影响很小。 而对于B, 当波长小于490 nm范围随浓度增加吸光度值逐渐下降, 超过490 nm波长范围吸光度值随浓度升高而升高, 这是由于硼酸盐在水溶液中的化学行为复杂存在多种溶液结构[16], 可能影响了十八胺和溴酚蓝的络合, 有待进一步研究证实。 对250~650 nm波长范围的吸光度进行相对误差计算, 当B的浓度达到50 mg· L-1时, 如图5(d)所示在这一波长范围内相对误差小于或接近于± 5%, 浓度超过50 mg· L-1后相对误差逐渐增大, 可通过多羟基化合物萃取预处理除硼, 消除或降低硼酸盐对测定的干扰[17]。
 | 图5 不同浓度微量组分对十八胺萃取体系吸收光谱的影响 (a): 锂离子溶液; (b): 铵根离子溶液; (c): 硼酸盐溶液; (d): 不同浓度硼酸盐溶液光谱曲线Fig.5 Effect of trace components with different concentrations on the absorption spectrum of octadecylamine extraction system (a): Lithium ion solution; (b): Ammonium ion solution; (c): Borate Solution; (d): Relative error of spectral curves at 250~650 nm for solutions with different borom concentrations |
平衡时间对十八胺萃取体系吸收光谱的影响如图6所示。 随着平衡时间延长, 小于490 nm范围波长光谱曲线基本保持不变, 结合2.2节讨论分析中在波段一和波段二范围情况及实际操作需要, 平衡时间为2 min即可。 对于大于490 nm波长范围0~2 min的光谱曲线下降较为明显, 2 min后下降幅度较小。 络合物氢键的强弱与氢键的给予体和接受体有关, 溴酚蓝分子上C原子电负性较小, 与十八胺形成的氢键作用弱(C— H…N), 在本实验中, 溶液反复受到光的照射, 推测由于光的反复照射导致了十八胺络合物氢键作用的削弱, 改变十八胺络合方式, 使此波段吸光度值下降。
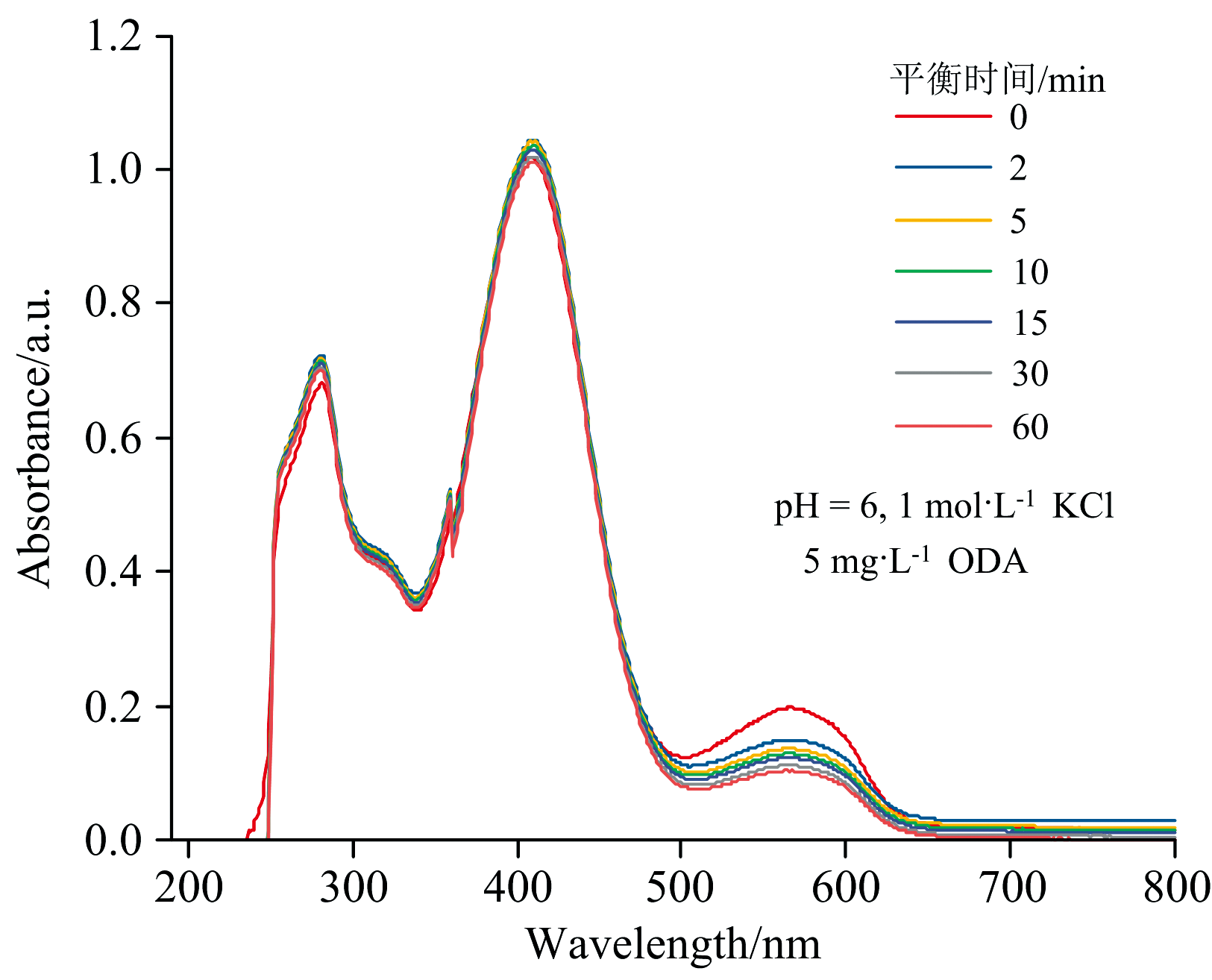 | 图6 平衡时间对十八胺萃取体系吸收光谱的影响Fig.6 Effect of equilibrium time on the absorption spectrum of octadecylamine extraction system |
由图7(a, b)中可以看出, 同一浓度下随着显色剂用量的增加, 空白组与实验组吸光度值均整体呈上升趋势, 且实验组405 nm附近峰高于280 nm附近的吸收峰, 且570 nm 处出现新峰, 表明溴酚蓝分子和十八胺络合物均被有机溶剂萃取。 实验组中, 显色剂用量为0.25 mL时溴酚蓝钠盐与十八胺摩尔比为1∶ 1, 溶液中十八胺可完全络合并萃取。显色剂用量增加到0.75 mL时溴酚蓝大量过量, 可能出现自消光现象, 导致吸光度值太大且不符合朗伯-比尔定律。 本研究选择0.5 mL的用量较为合适。
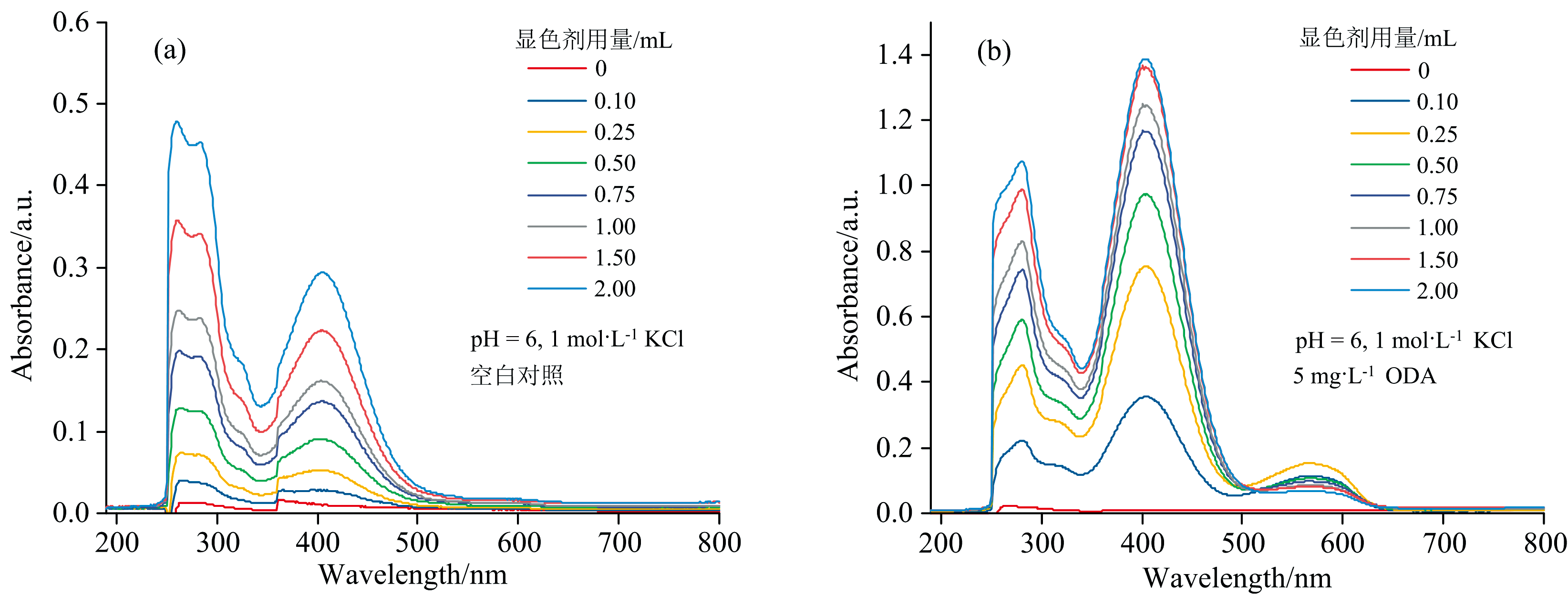 | 图7 不同显色剂用量对十八胺萃取体系吸收光谱的影响 (a): 0 mg· L-1十八胺溶液; (b): 5 mg· L-1十八胺溶液Fig.7 Effect of different chromogenic agent dosage on the spectrum of octadecylamine complex at octadecylamine solution of (a) 0 mg· L-1, (b) 5 mg· L-1 |
对所得到的不同浓度十八胺萃取体系吸收光谱进行了相关系数分析。 通过相关系数法计算样品浓度于吸光度两个向量之间的相关程度。 相关系数R由式(1)计算, 样品浓度值与吸光度的相关系数随波长变化曲线如图8(a)所示。
${{R}_{\lambda }}=\frac{\overset{n}{\mathop{\mathop{\sum }_{i=1}}}\,\left( {{x}_{i,\lambda }}-{{{{\bar{x}}}}_{\lambda }} \right)\left( {{y}_{i}}-{\bar{y}} \right)}{\overset{n}{\mathop{\mathop{\sum }_{i=1}}}\,\sqrt{{{({{x}_{i,\lambda }}-{{{{\bar{x}}}}_{\lambda }})}^{2}}{{({{y}_{i}}-{\bar{y}})}^{2}}}}$(1)
式(1)中, ${\bar{x}}=\frac{\overset{n}{\mathop{\mathop{\sum }_{i=1}}}\,{{x}_{i,\lambda }}}{n}$, ${\bar{y}}=\frac{\overset{n}{\mathop{\mathop{\sum }_{i=1}}}\,{{y}_{i}}}{n}$, λ =1, 2, 3, …, m, m为波长数, i=1, 2, 3, …, n, n为样品数。
 | 图8 (a)不同浓度十八胺萃取体系吸收光谱, (b)样品浓度与吸光度的相关系数随波长变化曲线, (c)和(d)相关系数和摩尔吸光系数随波长的变化曲线Fig.8 (a) Absorption spectrum of octadecylamine extraction system with different concentrations, (b) curve of correlation coefficient between sample concentration and absorbance versus wavelength, (c) and (d) variation curve of correlation coefficient and molar absorption coefficient with wavelength |
图8(a)中样品浓度与吸光度的相关系数随波长变化。 对190~800 nm波长范围进行了相关系数分析计算, 图8(b)中, 当波长范围在324~329 nm相关系数R均大于0.998 6, 最接近于1。 结合2.2节, 波段一为329~381 nm波长范围内, 在离子强度为1 mol· L-1时对吸光度值影响较小。 排除波长范围在324~329 nm中建立测定工作曲线的选择。 波段二在453~470 nm的波长范围, 相关系数和摩尔吸光系数分别表示评估不同波长下工作曲线的精密度和灵敏度。 如图8(c)所示的相关系数和摩尔吸光系数在波段二的变化曲线, 通过对工作曲线精密度和灵敏度的控制可选择不同波长建立工作曲线。 由于测定溶液中十八胺的浓度含量较低, 将摩尔吸光系数控制在1.20× 104 L· mol-1· cm-1以上以提高工作曲线的灵敏度, 当相关系数控制在0.994 0以上时, 根据图8(d)中交点可知, 458 nm处建立的工作曲线精密度和灵敏度较为合适。
如图9工作曲线所示, 十八胺含量在0~10 mg· L-1范围内时溶液吸光度与浓度呈良好线性关系, 线性拟合方程为A=0.049 49c+0.066 24, 相关系数R2为0.992 3, 摩尔吸光系数ε 为1.33× 104 L· mol-1· cm-1。 为确定检出限, 连续20次测定空白溶液的吸光度, 吸光度的标准偏差σ =1.40× 10-2, 根据IUPAC推荐的检出限公式D.L.=3σ /r (r为标准曲线的斜率), 计算出该分析方法的最低检出限为0.85 mg· L-1。
 | 图9 工作曲线Fig.9 Working curve |
为了考察方法的精密度, 配制系列不同浓度的十八胺溶液, 按照1.4.5节中加入缓冲溶液和显色剂, 于458 nm处测定吸光度, 每个样平行测定6次。
如表1所示, 此方法测定十八胺溶液吸光度的相对标准偏差为0.33%~2.63%。
| 表1 测定十八胺溶液浓度的重复性 Table 1 Repeatability of determination of octadecylamine concentration |
配制系列不同浓度十八胺溶液, 按照1.4.5节中加入缓冲溶液、 显色剂及萃取剂, 于458 nm处测定吸光度。 结果如表2所示, 测量浓度与样品浓度的相对误差在-7.50%~+3.00%之间, 平均相对误差为-0.90%。
| 表2 测定氯化钾溶液中十八胺的浓度及相对误差 Table 2 Determination of concentration and relative error of octadecylamine in potassium chloride solution |
配制pH为6的10、 25和40 mg· L-1十八胺溶液和25 mg· L-1十二烷基吗啉溶液, 分别取5 mL三组不同浓度的十八胺溶液于25 mL容量瓶中, 各加入5 mL 25 mg· L-1十二烷基吗啉溶液, 按照1.4.5节加入缓冲溶液、 显色剂及萃取剂, 于458 nm处测定吸光度。 结果如表3所示, 测量浓度与样品浓度的平均相对误差在-0.25%。
| 表3 测定十八胺和十二烷基吗啉混合溶液中十八胺的浓度 Table 3 Determination of octadecylamine concentration in mixed solution of octadecylamine and dodecylmorpholine |
采用汞量法测定出正浮选精钾母液Cl-浓度为5.52 mol· L-1, 取4.5 mL正浮选精钾母液于25 mL容量瓶中, 使离子强度为1 mol· L-1氯化钾当量, 并向正浮选精钾母液中加入不同体积pH为6的18 mg· L-1十八胺溶液, 按照1.4.5节加入缓冲溶液、 显色剂及萃取剂, 于458 nm处测定吸光度。 结果如表4所示, 正浮选精钾母液的浓度为8.66 mg· L-1, 加标回收率为95.5%~106%。
| 表4 样品分析及加标回收率 Table 4 Sample analysis and spiked recovery |
十八胺与溴酚蓝通过范德华力和氢键作用形成络合物, 乙酸丁酯可萃取十八胺-溴酚蓝络合物, 通过调控溶液pH可避免十二烷基吗啉对测定的干扰。 以乙酸丁酯为萃取剂、 溴酚蓝钠盐为显色剂, 采用分光光度法测定氯化钾中十八胺的浓度, 溶液的吸光度与其浓度在0~10 mg· L-1内遵守朗伯-比尔定律。 在波长为458 nm处测定方程为A=0.049 49c+0.066 24, 相关系数R2为0.992 3, 测量十八胺浓度的相对标准偏差为0.33%~2.63%, 平均相对误差为-0.90%。 测量十八胺和十二烷基吗啉混合体系的平均相对误差为-0.25%, 测得正浮选精钾母液的浓度为8.66 mg· L-1, 加标回收率为93.1%~111%, 可为正浮选工艺研究和钾盐纯化提供便捷可靠的测试方法。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|