

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
纳米Ce1-4 x(FeAlCoLa) xO2- δ固溶体微观光谱特征及氧化还原性能研究
[孙世龙1  , 张国芳
, 张国芳1, * , 束俊1 , 郭瑞华1 , 李一鸣1 , 刘卓承1 , 许剑轶1 , 葛启录2 ]
, 张国芳, 束俊|
|


作者简介: 孙世龙, 2000年生,内蒙古科技大学材料与冶金学院材料化学系硕士研究生 e-mail: 1185666716@qq.com
采用水热法合成Fe3+、 Al3+、 Co2+及La3+共掺杂纳米Ce1-4 x(FeAlCoLa) xO2- δ( x=0.00~0.05)固溶体, 利用X射线衍射(XRD)、 透射电子显微镜(TEM)、 扫描电子显微镜(SEM)、 紫外吸收光谱(UV)、 荧光光谱(PL)、 拉曼光谱(Raman)以及与H2的程序升温还原反应(TPR)等方法对固溶体的微观结构、 形貌、 光谱特征和氧化还原活性进行系统表征及分析。 XRD结果表明, Ce1-4 x(FeAlCoLa) xO2- δ固溶体均呈CeO2立方萤石结构, 当掺杂量增加到 x=0.04时, 在36.6°处出现了微弱的Co3O4杂相, 可以确定掺杂离子在CeO2晶格中的固溶度 x<0.04。 样品的(111)衍射峰位向高角度偏移, 表明掺杂离子引起晶格发生畸变。 TEM及SEM结果显示样品为球形纳米颗粒, 掺杂离子引起晶面间距变小。 紫外吸收光谱表明, 与纯CeO2相比, 掺杂样品的吸收边逐渐红移, 在560~780 nm范围观察到掺杂离子的紫外吸收峰。 掺杂引起样品能隙降低, 从2.84 eV(纯CeO2)逐渐降低至2.10 eV( x=0.05)。 其原因可归结为掺杂离子在CeO2的价带和导带之间形成新的杂质能级, 允许电子从价带跃迁到较低的杂质能级上, 继而降低了跃迁能隙。 由于掺杂离子引起晶格内部发生畸变以及氧空位比例增大, 阻碍了电子的高能跃迁, 也可引起能隙减小。 荧光光谱证明, 掺杂样品的发射峰强度明显降低。 Raman光谱表明, 掺杂引起F2g峰位发生偏移, 峰强减小, 峰宽变大。 同时, 对应于氧空位峰的相对强度逐渐提高。 荧光光谱及Raman光谱均证明掺杂离子引起固溶体晶格畸变程度增加, 氧空位浓度提高。 H2-TPR测试表明, 掺杂可以有效降低CeO2的氧化还原反应温度, 提高氧化还原活性, 当 x=0.03的样品表面还原温度最低, 还原峰的面积最大, 即氧化还原反应活性最佳, 表明样品的氧化还原性能与晶粒尺寸、 晶格缺陷及氧空位浓度密切相关。 通过以上研究证明, 四种离子共掺杂CeO2能够有效修饰微观晶体结构, 在较低掺杂浓度下即可显著改善样品的催化活性。
, ZHANG Guo-fang, SHU JunHydrothermal method was used to synthesize nanosized Fe3+, Al3+, Co2+, and La3+ co-doped Ce1-4 x(FeAlCoLa) xO2- δ( x=0.00~0.05) solid solutions. The solid solutions’ microstructure, morphology, spectral characteristics, and redox activities were systematically characterized and analyzed by XRD, TEM, SEM, UV, PL, Raman, and temperature-programmed reduction (TPR) with H2. XRD results showed the Ce1-4 x(FeAlCoLa) xO2- δ solid solutionsexhibited the CeO2 cubic fluorite structure. A tiny diffraction peak corresponding to the Co3O4 impurity phase at 36.6°was observed when the doped content reached x=0.04, indicating that x=0.04 was the solid solubility of doped ions in the CeO2 lattice. The positions of the (111) diffraction peaks were shifted towards a higher angle, which proved the doped ionsinduced the distortion of the lattice. The TEM and SEM images showed the samples were spherical with high crystallinity, and doping caused lattice contraction. The UV absorption spectra revealed that the doped samples’ absorption edges were gradually red-shifted compared to pure CeO2. Extra absorption peaks corresponding to the doped ions were found in the region of 560~780 nm. The band gap energies decreased from 2.84 eV (pure CeO2) to 2.1 eV ( x=0.05). The reasoncould be that the doped ions formed new impurity energy levels between the valence and conduction bands, which allowed the electrons to transition from the valence band to the lower impurity energy levels and then lowered the band gap energies. In addition, the distortion of the lattice and increased concentration of oxygen vacancies prevented the electrons from transferring to higher energies, which can also result in the reduction of band gap energies. PL spectra showed that doping significantly reduced the emission peak intensities. Raman spectra demonstrated that the dopingresulted in the shift of the F2g peak, the decrease of peak intensities, and the widening of peaks. Meanwhile, the relative intensities of the peak corresponding to the oxygen vacancies were also observed to be enhanced. Thus, both the PL and Raman spectra proved that doping increased the degree of lattice distortion and the concentration of oxygen vacancies. The H2-TPR test results showed that doping can effectively reduce the redox reaction temperatures and improve the redox activities. The sample doped with x=0.03 possess the lowest surface reduction temperature and the largest peak areas, which meansthis sample exhibited the best redox activities. It can be concluded that the redox performances of the samples were closely related to the grain sizes, lattice defects, and oxygen vacancy concentrations. This study showed that the four ions co-doped with CeO2 could effectively modify the microstructure and improve the samples’ catalytic activities at a low doping concentration.
CeO2因具有优越的氧化还原性能而得到广泛应用。 在氧化还原过程中, CeO2中的Ce4+易还原为Ce3+, 作为电荷补偿, 会伴随晶格内氧原子的去除, 由此形成的氧空位可在催化过程有效提升其活性[1]。 但是纯CeO2的催化活性和氧空位浓度较低, 因此如何提高CeO2的反应活性成为了研究热点。 改善CeO2的性能主要包括以下方法: 一是提高比表面积, 增加表面活性点密度, 从而改善CeO2的性能; 二是通过在CeO2晶格中掺杂其他离子形成固溶体, 掺杂可引起电荷补偿而增加晶格内氧空位浓度, 达到优化CeO2的催化性能及反应活性的目的[2]。 制备方法也会影响样品的性能, 水热法具有流程简单、 制得的粉体粒度均匀、 分散性较好、 纯度高等优点, 因此成为一类极具优势的制备纳米材料的方法[3]。
目前关于掺杂CeO2的研究很多, 包括掺杂过渡金属离子, 主族金属离子以及稀土金属离子等。 近年来, 关于多离子共掺杂CeO2所引起的协同效应也逐渐引起人们的关注。 研究表明, Fe3+可以有效降低CeO2的带隙[4], Al3+能够明显增加CeO2的氧空位浓度[5], Co2+可调节CeO2的紫外带隙, 提高催化活性及稳定性[6], La3+能够促进CeO2晶格中氧空位的形成, 从而提高催化剂的活性[7]。 综合以上研究可知, 掺杂Fe3+、 Al3+、 Co2+和La3+都可以在不同方面有效改善CeO2的氧化还原性能。 如果能够将以上几种效应综合起来, 相信掺杂CeO2固溶体的反应活性能够得到进一步提升。 为研究Fe3+、 Al3+、 Co2+和La3+共同掺杂对CeO2的影响, 本文采用水热法制备纳米Ce1-4x(FeAlCoLa)xO2-δ固溶体, 系统研究四离子同比例掺杂对CeO2的微观结构、 光谱特征及氧化还原活性等方面的影响机制。
水热法制备纳米Ce1-4x(FeAlCoLa)xO2-δ固溶体所需药品包括Ce(NO3)3·6H2O (AR)、 La(NO3)3·6H2O (AR)、 Al(NO3)3·9H2O (AR)、 Fe(NO3)3·9H2O (AR)、 Co(NO3)2·6H2O (AR)。 首先将所需硝酸盐分别配制为0.3 mol·L-1的溶液, 按照掺杂比例取适量溶液充分混合, 在搅拌的同时逐滴加入氨水, 调节溶液pH约为8。 所得悬浊液充分反应后, 移入以聚四氟乙烯为内衬的不锈钢高压反应釜中, 200 ℃水热反应24 h。 水热反应完成后将样品用蒸馏水充分洗涤抽滤, 80 ℃下干燥6 h。
利用射线源为CuKα1的X射线衍射仪(D/Max-2400)检测样品的晶体结构, 扫描范围20°~80°, 扫描速度4°·min-1; 分别利用透射电子显微镜(JEM2100)及场发射扫描电子显微镜(Sigma-500)来观察样品的微观形貌; 样品的紫外吸收光谱利用紫外可见分光光度仪(Hitachi U-3900)进行测试, 测试范围为200~800 nm, 扫描速率为300 nm·min-1; 荧光光谱利用荧光分光光度计(Hitachi, F-4600)进行测试, 激发波长为325 nm; Raman光谱利用激光Raman光谱仪测试(Horiba, JY-HR800), 测试范围为50~1 200 cm-1。 样品的氧化还原性能(H2-TPR)的测试是通过化学吸附仪(彼奥德, PC-1200)完成, 采用的气体为质量比为H2(10%)-N2(90%)的混合气体, 测试温度范围为室温至900 ℃, 升温速率为10 ℃·min-1。
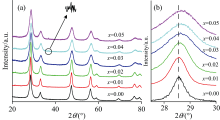
图1(a)为样品的XRD谱图, 由图可知, 纯CeO2及其掺杂样品的衍射峰与CeO2标准谱(PDF#34-0394)相匹配, 当掺杂离子浓度x=0.01~0.03时, 样品的XRD图谱显示为CeO2纯相, 表明Fe3+、 Al3+、 Co2+和La3+进入到CeO2萤石立方晶格中形成了CeO2固溶体。 而掺杂比例为x=0.04和0.05的样品中观察到位于36.6°的微弱杂峰(如箭头所示), 该峰对应于Co3O4的衍射峰[8]。 因此, 可将四种掺杂离子在CeO2中的固溶限初步确定为x<0.04。 图1(b)为样品(111)晶面的衍射峰放大图, 随着掺杂浓度增加, 衍射峰强度逐渐降低, 峰宽变大, 表明掺杂会降低样品晶粒的结晶度[9], 同时峰位也逐渐向高角度偏移, 说明掺杂会引起晶格发生畸变。
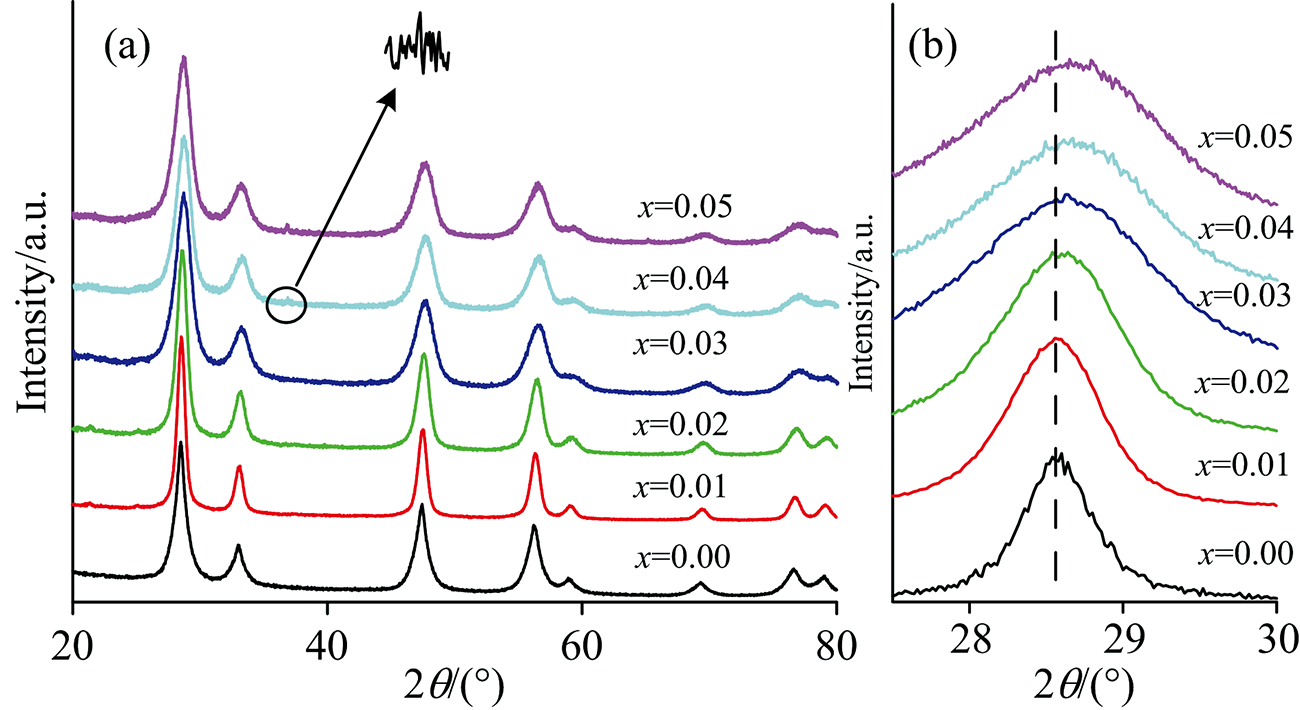 | 图1 Ce1-4x(FeAlCoLa)xO2-δ固溶体的(a)XRD图; (b) (111)晶面衍射峰放大图Fig.1 (a) XRD patterns and (b) enlarged (111) plane diffraction peaks of Ce1-4x(FeAlCoLa)xO2-δ solid solutions |
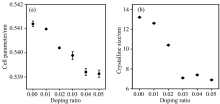
通过对XRD衍射图谱进行Rietveld精修来确定样品的晶格常数, 结果如图2(a)所示, 随着掺杂量的增加, 样品的晶格常数逐渐减小。 分析认为Fe3+(0.78 Å , Ⅷ 配位)、 Al3+(0.535 Å , Ⅵ 配位)、 Co2+(0.9 Å , Ⅷ 配位)离子的半径均小于Ce4+(0.97 Å , Ⅷ 配位)离子, 当掺杂离子进入CeO2晶格内, 小半径的掺杂离子替代Ce4+的晶格位置导致晶格发生收缩, 虽然La3+(1.16 Å , Ⅷ 配位)离子半径稍大于Ce4+, 但不影响晶格常数减小的整体趋势。 与x=0.04样品相比, x=0.05样品的晶格常数只是略有减小, 分析认为当掺杂量达到0.04后出现Co3O4杂相, 表明CeO2晶格内无法容纳更多的Co2+, 此时继续掺杂只有 Fe3+及 Al3+两种离子引起晶格常数的收缩, 因此晶胞的收缩程度不再明显变化。 通过Scherrer公式计算样品的晶粒尺寸, 结果如图2(b)所示。 随掺杂浓度的提高, 样品的晶粒尺寸逐步减小。 其中纯CeO2的晶粒尺寸为13.2 nm, 掺杂浓度x=0.05时的晶粒尺寸为6.8 nm, 说明掺杂离子可阻止样品晶粒长大[1]。
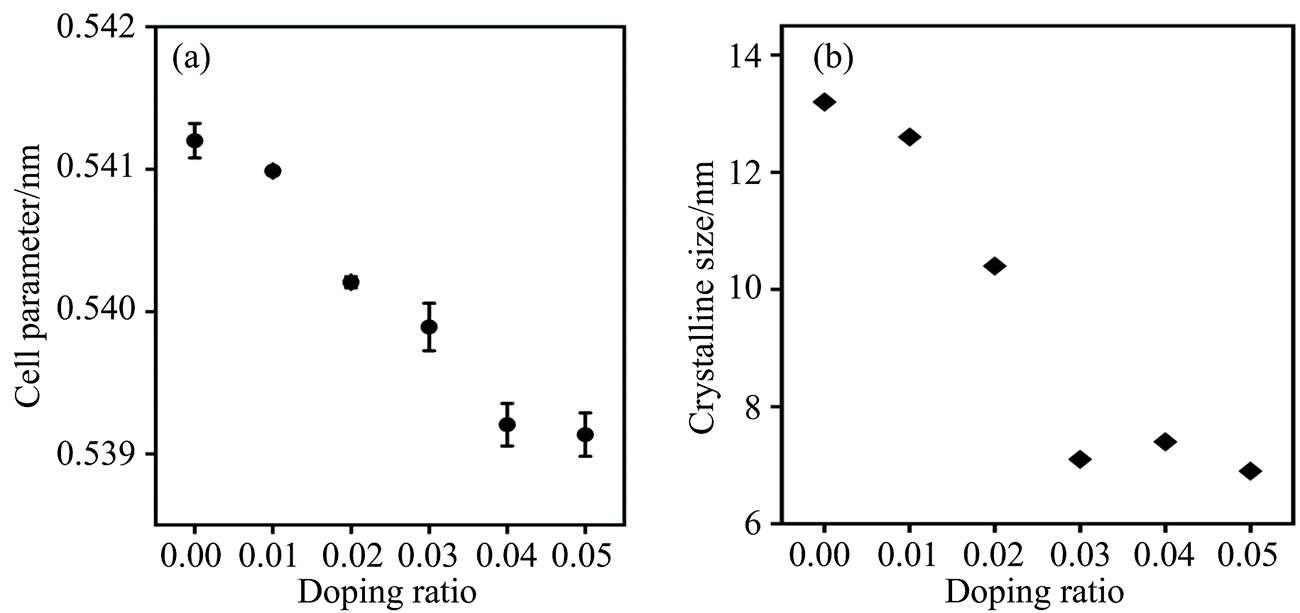 | 图2 Ce1-4x(FeAlCoLa)xO2-δ(x=0.00~0.05)的(a)晶格常数; (b) 晶粒尺寸Fig.2 (a) Lattice parameters and (b) crystalline sizes of Ce1-4x(FeAlCoLa)xO2-δ(x=0.00~0.05) |
图3(a)为典型样品Ce0.88(FeAlCoLa)0.03O2-δ的TEM结果, 样品的晶粒尺寸分布较为均匀, 大小约10 nm。 图3(b)为该样品的高分辨图, 插图为局部放大图, 样品晶面条纹清晰, 表明样品结晶度良好。 测量得到晶面间距为0.299 nm, 确定该晶面为(111)晶面, 纯CeO2晶面间距为0.312 nm, 证明小半径的离子掺杂引起晶格收缩, 该结果与XRD的分析相一致。 通过SEM进一步观察Ce0.88(FeAlCoLa)0.03O2-δ样品的形貌特征, 如图3(c)所示, 可以清晰地观察到该样品呈现为粒径大小均匀的球形颗粒。
 | 图3 Ce0.88(FeAlCoLa)0.03O2-δ的(a) TEM图; (b)高分辨图; (c) SEM图Fig.3 (a) TEM diagram; (b) high-resolution map; and (c) SEM diagram of Ce0.88(FeAlCoLa)0.03O2-δ |
图4为样品的紫外吸收光谱, 通过图4(a)可以观察到样品的紫外吸收光谱均在200~350 nm处存在强吸收带, 其中240 nm的吸收峰为从O2+跃迁到Ce3+的电荷转移, 260和330 nm吸收峰对应于从O2+到Ce4+的电荷转移及带间跃迁[9, 10]。 与纯CeO2样品对比, 掺杂样品在560~780 nm范围内均出现一个宽吸收峰, 且随着掺杂浓度的提高, 强度逐渐增大, 表明该区域存在掺杂离子的紫外吸收峰, 拟合发现该宽峰对应为Fe3+和Co2+的吸收峰[11, 12]。 图4(b)为通过固溶体的紫外吸收光谱按照直接跃迁模式拟合所得能隙图, 附图为能隙值与掺杂量的变化关系。 由附图可知, 随着掺杂离子浓度的增大, 紫外能隙值逐渐降低, 表明掺杂能够使CeO2的能隙明显减小。 纯CeO2样品的能隙值为2.84 eV, 当掺杂量x=0.05时, 能隙值降低至2.1 eV。 能隙降低的主要原因是掺杂离子可在CeO2的价带和导带之间形成新的杂质能级, 允许电子从价带跃迁到较低的杂质能级上, 继而降低了跃迁能隙, 导致带隙能量减小; 掺杂离子能够提高样品晶格内的氧空位浓度及晶格畸变程度, 使分子内电荷分布发生改变, 阻碍电子的高能跃迁, 从而减小能带隙[13]。 从图4(b)还可以观察到, 当掺杂浓度从0.04升高至0.05后, 样品的能隙降低趋势减缓, 分析认为此时固溶体中Co2+浓度不再增加, 此时仅有三种离子(Fe3+、 Al3+及La3+)继续对能隙起作用, 因此导致能隙减小的幅度降低。
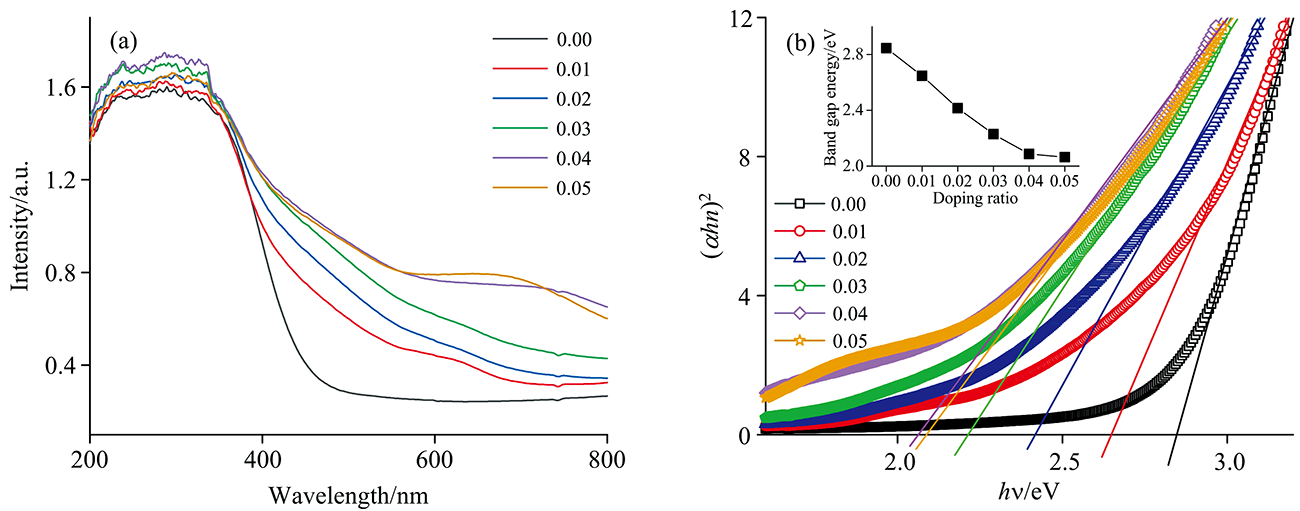 | 图4 (a) Ce1-4x(FeAlCoLa)xO2-δ的紫外吸收光谱; (b)紫外能隙图, 附图为能隙与掺杂量关系Fig.4 (a) UV absorption spectra of Ce1-4x(FeAlCoLa)xO2-δ; (b) UV energy gap diagram with accompanying plot of energy gap versus doping amount |
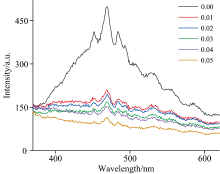
荧光光谱可反映晶格内受激电子和空穴的复合情况, 可以用来表征固溶体晶格内微观结构及环境的变化。 图5为样品的荧光光谱图。 所有样品的荧光发射峰的形状相似, 但各样品的峰强度差别很大。 468 nm处的发射峰为相对最强的信号峰, 而其他几个较弱的峰分别位于410、 425、 451、 484及505 nm处等, 这些峰相互叠加, 共同组成了400~600 nm范围的发射宽峰。 这些峰是CeO2中O(2p)和Ce(4f)能级之间存在的晶格缺陷所构成的能带向O(2p)的跃迁所引起的发射峰[9]。 通过对比各样品的峰强度, 纯CeO2(x=0.00)的发射峰强度最大, 表明电子和空穴对能够以较高比例复合。 当掺杂离子被引入晶格后, 峰值强度骤降, 分析认为掺杂导致CeO2晶体结构发生畸变, 并产生大量氧空位, 从而延迟受激电子和空穴的复合过程[13], 引起峰的强度明显降低。 随着掺杂浓度的增加, 发射峰的强度持续降低, 表明样品中的晶格缺陷及氧空位浓度随掺杂浓度而升高, 尽管当掺杂浓度为0.04时固溶体中出现了Co3O4杂相, 但其他三种离子(Fe3+、 Al3+及La3+)仍可以继续掺入固溶体中, 使固溶体的晶格缺陷及氧空位占比继续增大, 峰的强度持续降低, 该结论与 XRD和紫外分析结果一致。
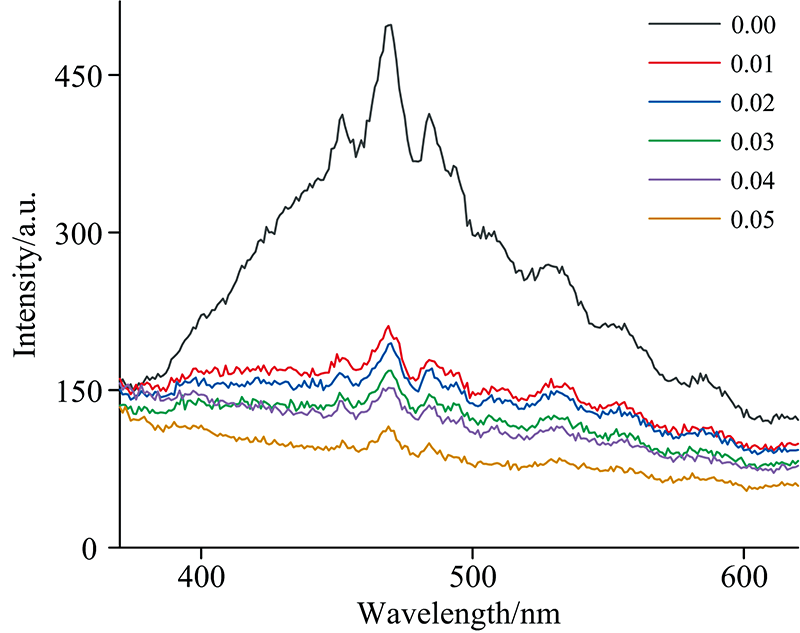 | 图5 Ce1-4x(FeAlCoLa)xO2-δ固溶体的荧光光谱Fig.5 PL spectra of Ce1-4x(FeAlCoLa)xO2-δ solid solutions |
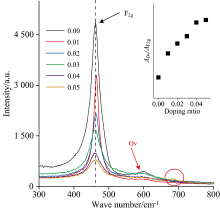
图6为Ce1-4x(FeAlCoLa)xO2-δ的Raman图谱, 可以观察到, 各样品均在465 cm-1附近有一个振动强峰, 该峰为萤石立方结构CeO2晶格中Ce4+与其附近的O2-所产生的F2g模式对称振动[9]。 对比不同掺杂浓度样品的F2g振动峰, 可以发现, 随掺杂浓度增加, F2g峰位向低波数偏移, 从465 cm-1(x=0.01)逐渐偏移至458 cm-1(x=0.05), 同时, 掺杂离子还引起F2g振动峰强度逐渐降低, 峰宽逐渐变大, 对称性降低。 峰位向低波数移动主要是由于掺杂使样品的晶格参数减小, 改变了晶格内原子间作用力而引起的[14]。 另一方面, 掺杂引起样品晶粒尺寸减小, 晶格缺陷浓度增加, 导致峰的强度降低及峰宽变大[15]。 在600 cm-1处观察到一个强度较弱的宽峰(如红色箭头所示), 对应于氧空位特征峰(Ov)[16], 通常该峰相对强度越高, 则代表氧化物晶格中氧空位浓度越高。 通过Ov和F2g的峰面积比值(
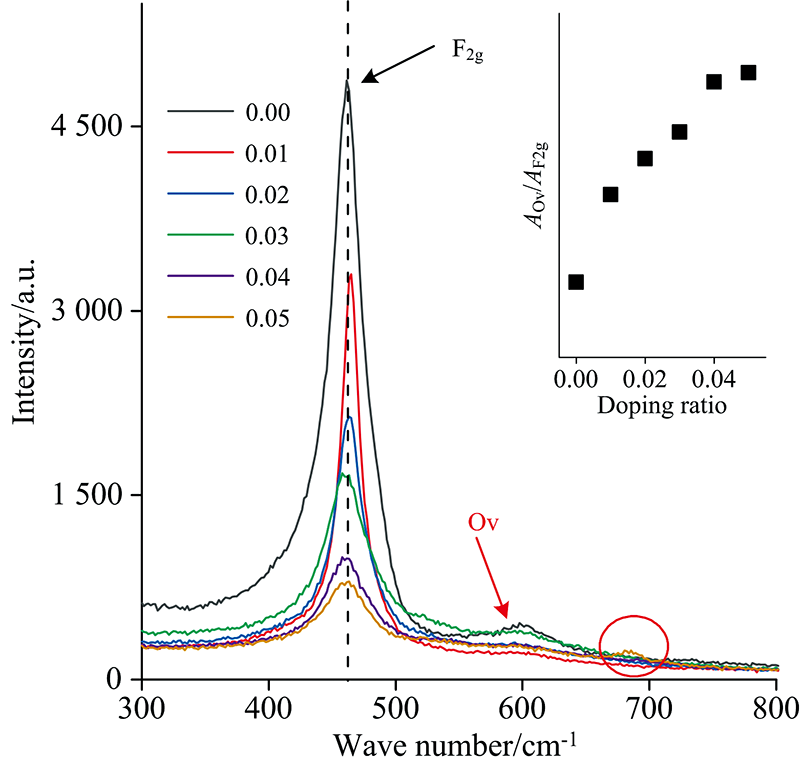 | 图6 Ce1-4x(FeAlCoLa)xO2-δ的Raman图谱, 附图为 |
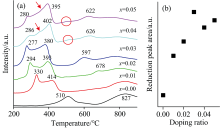
图7(a)为Ce1-4x(FeAlCoLa)xO2-δ的程序升温还原(TPR)曲线。 纯CeO2第一个还原峰的峰值位于510 ℃, 对应于CeO2的表面还原峰; 第二个峰值位于827 ℃, 代表CeO2的体相还原温度[9]。 与纯CeO2相比, 掺杂样品的表面还原峰劈裂为两个峰, 且还原温度明显降低。 其中掺杂浓度0.01~0.03样品的第一个表面还原温度逐渐降低, 从330 ℃逐渐降为277 ℃, 而掺杂浓度0.04及0.05样品的表面还原温度稍有升高, 分别为286及280 ℃。 第二个表面还原峰的温度从414 ℃逐渐降至380 ℃, 随后又略微上升。 结合XRD结果可知, 固溶体掺杂浓度达到0.04时, 样品中出现了Co3O4杂相[18], 因此掺杂样品表面还原温度增高的原因可归结为表面存在Co3O4杂相[20]。 与纯CeO2相比, 掺杂样品的表面还原峰数目变多, 这是由于样品中掺杂了四种金属离子, 导致样品氧空位浓度显著增加, 促使更多体相中的Ce4+转移到表面[19], 同时, 掺杂引起样品晶粒尺寸变小, 比表面积增大, 也进一步增加了样品表面Ce4+的量, 因此表面还原过程分为两步进行。 低温处的表面还原过程, 可归因于CeO2样品表面羟基的还原[20], 而出现在稍高温度的第二个表面还原峰, 可归结为CeO2表面氧与H2的还原反应, 使得Ce4+变为Ce3+。
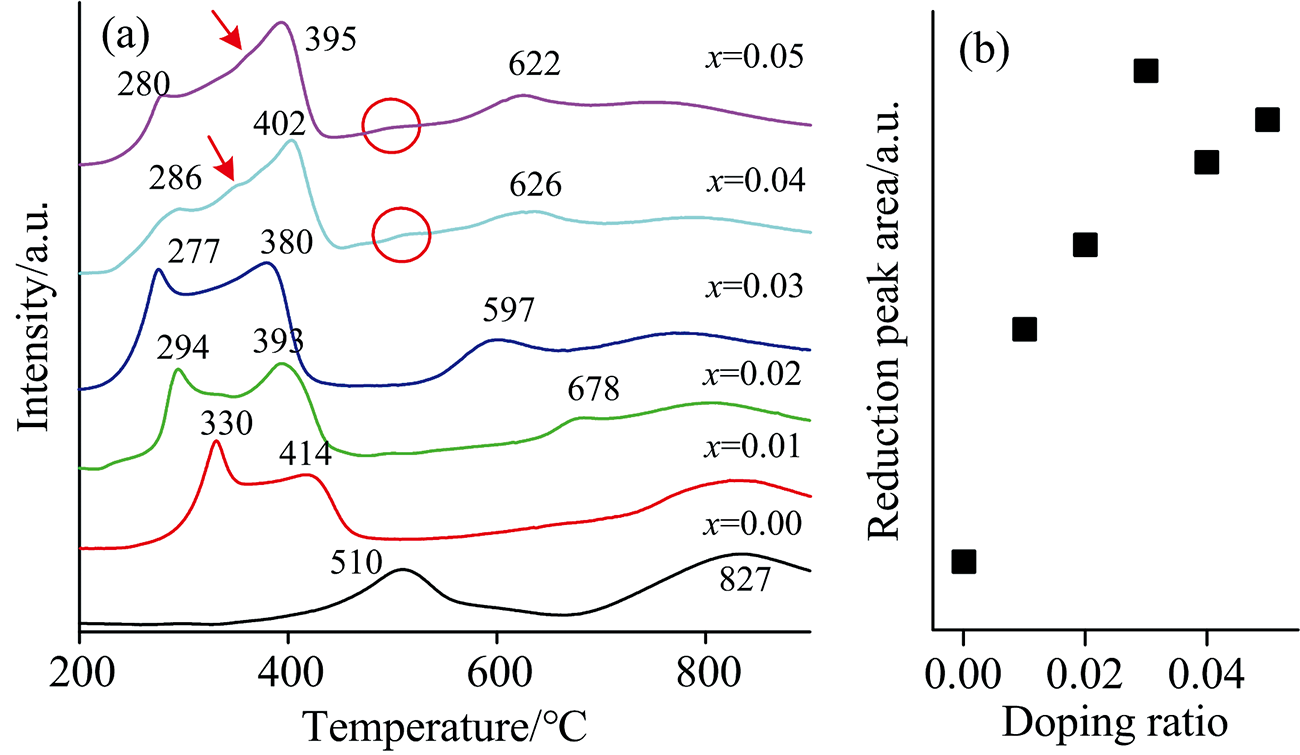 | 图7 (a) Ce1-4x(FeAlCoLa)xO2-δ的H2-TPR曲线; (b) 还原峰面积变化曲线Fig.7 (a) H2-TPR curves of Ce1-4x(FeAlCoLa)xO2-δ; (b) Reduction the peak area curve |
由于掺杂离子使得表面还原不仅有Ce4+的还原过程, 还有多价态金属离子的还原, 使其还原过程变得复杂, 出现一些新的还原峰。 当x=0.04~0.05时, 在还原峰温度较低一侧的肩峰, 即位于350 ℃处的微弱还原峰(如箭头所指), 可归结为样品表面高度分散的Co3O4的还原峰[20]。 在500 ℃处出现微弱的还原峰(如图圈处), 对比氧化铁的峰位分析后, 确定该峰为氧化铁的还原峰[21]。 另外在x=0.02~0.05时, 在出现678、 597、 626和622 ℃处较强的还原峰, 该还原峰可归因于体相还原峰的偏移, 同时也可能存在Al3+的还原[22]。
样品消耗H2量的大小可以通过还原峰的面积大小进行定性比较, 结果如图7(b)所示。 通过对比, 纯CeO2的峰面积最小, 随着掺杂浓度的提高, 峰面积显著增大。 当掺杂浓度为0.03时, 峰面积达到最大, 而掺杂浓度为0.04~0.05时, 还原峰面积又有减小的趋势。 当掺杂浓度为0.00~0.03时, 随着掺杂浓度的提高, 氧空位逐渐增大, 因此还原峰面积也随之而增大。 但当掺杂浓度超过0.03后, 固溶体表面出现了高度分散的Co3O4杂相, 因纯Co3O4的程序升温还原温度更高[20], 导致固溶体在与H2的还原反应过程中, 表面还原受到阻滞, 还原温度升高。 此外, 杂相的存在还会影响到固溶体与H2的反应量, 进而使表面还原峰面积降低。
掺杂的四种金属离子能够有效地增大固溶体的氧空位浓度及晶格缺陷, 降低还原起始温度及峰值温度, 使得固溶体的氧化还原活性得到显著提升。 其中, x=0.03的样品表面还原温度最低, 该样品的峰面积最大, 即氢反应活性最大。
水热法合成纳米Ce1-4x(FeAlCoLa)xO2-δ固溶体。 系统研究了固溶体的微观结构、 光谱特征和H2反应活性。
(1) 四种离子在CeO2中的固溶限x<0.04。 随着掺杂浓度的增加, 固溶体结晶度降低, 晶格畸变加剧, 晶格常数降低。
(2) 掺杂样品的紫外吸收光谱发生红移, 能隙降低; Raman光谱及荧光光谱证明了掺杂离子引起CeO2中氧空位和缺陷含量增加。
(3) TPR测试发现x=0.03的样品表面还原温度最低, 峰面积最大, 与氢反应活性最大。 证明了氢反应活性的影响因素与晶格常数、 晶粒尺寸、 缺陷和氧空位浓度及样品的表面状态有关。 进一步证明在四种离子共掺杂的协同作用下, 能够有效修饰晶格, 提高反应活性。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|