
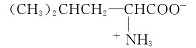

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
亮氨酸与异亮氨酸太赫兹光谱的振动模式分析
[刘晓松1, 2  , 赵国忠
, 赵国忠1, * , 屈媛2 ]
, 赵国忠, 屈媛|
|


作者简介: 刘晓松, 1998年生, 内蒙古大学物理科学与技术学院硕士研究生 e-mail: liuxiaosong0476@163.com
氨基酸是含有碱性氨基和酸性羧基的有机化合物, 是构成蛋白质的基本单位, 其种类, 数量和排列直接影响蛋白质的生物功能, 对维持机体功能有重要意义。 氨基酸分子间振动模式(扭转, 氢键和集体振动)大部分处于太赫兹(THz)波段, 表现出独特的吸收特征, 因此, 对氨基酸进行THz光谱研究, 能够更全面了解生物特性。 总结前人实测的亮氨酸与异亮氨酸位于0.2~2.6 THz波段的吸收谱, 同时, 利用量子化学计算方法解释其形成机理。 使用Gaussian09软件对单分子构型模拟计算, 模拟方法为半经验法(PM6), 从头计算法(HF, MP2)和密度泛函理论(B3LYP, M06-2X)结合6-311+G(d, p)高斯型基组; 使用Materials Studio 2019软件对晶胞构型模拟计算, 模拟方法为广义梯度近似的PBE, PBEsol, RPBE和WC等四种密度泛函结合平面波基组。 结果表明: 单分子构型模拟均缺少吸收峰位, 不同方法对同一振动模式的峰位计算不同, 因此, 对分子间相互作用较强的结构, 进行单一方法的该构型模拟, 很大程度不能正确匹配振动模式, 且受原子轨道线性组合方法影响, 与输入结构相比, 输出结构由COO-和$NH_{3}^{+}$基团变为COOH和NH2, 无法体现实际振动模式; 晶胞构型模拟对分子内和分子间振动模式描述, 吸收峰位与实测值匹配较好, 不存在质子转移情况, 较好指认实测峰位的振动模式。 亮氨酸与异亮氨酸使用PBEsol泛函计算结果最接近实测值, 说明模拟计算需充分考虑结构与泛函的匹配性, 即对结构交换关联能的描述, 也说明同一泛函对异构体的普适性, 此外, 不能以结构优化后差异作为判断泛函是否适用的标准。 晶胞构型计算结果包含分子间振动模式, 是单分子构型无法得到的结果, 且数据进行半峰全宽拟合, 导致两种构型结果在某一实测峰位处的振动模式存在差异。
As organic compounds containing an alkaline amino group and acidic carboxyl group, an amino acid is the basic unit of protein, whose type, quantity and arrangement directly affect the biological function of protein with great significance to maintain the body function. Most amino acid intermolecular vibrational modes (torsion, hydrogen bonding and collective vibrations) occur in the THz band and exhibit unique absorption characteristics. Consequently, THz spectroscopic studies of amino acids can give a more comprehensive understanding of biological properties. It was a summary of the absorption spectra of leucine and isoleucine located in the 0.2~2.6 THz band measured by previous authors, and, at the same time, the formation mechanism was explained using quantum chemical calculations. Using Gaussian09 software for single-molecule configuration simulations, the simulations were performed by semi-empirical method (PM6), ab initio method (HF, MP2) and density functional theory (B3LYP, M06-2X) combined with 6-311+G(d,p) Gaussian basis groups. Materials Studio 2019 software was used to calculate the cell configuration simulations for four density generalized gradient approximations such as PBE, PBEsol, RPBE and WC combined with plane wave basis groups. The results indicated that the single-molecule configuration simulations lacked absorption peak positions, and the peak positions were calculated differently for the same vibrational mode by different methods. So, for structures with strong intermolecular interactions, the simulation of this configuration by a single method did not correctly match the vibrational modes to a large extent and was influenced by the linear combination of atomic orbitals method. Compared to the input structure, the output structure changed from COO- and $NH_{3}^{+}$ groups to COOH and NH2, which did not reflect the actual vibrational mode. The intra and intermolecular vibrational modes were described by the cell configuration simulation. The absorption peak positions matched well with the measured values without proton transfer. The vibrational modes of the measured peak positions were better identified. The closest results of leucine and isoleucine calculations using the PBEsol general function to the measured values indicated that full consideration needs to be given to the matching of structure and general function in the simulation, i.e., the description of the structure exchange association energy and showed the universality of the same general function for the isomers. In addition, the difference after structural optimization cannot be used as the criterion to judge functional applicability. The results of cell configuration calculation include the intermolecular vibration mode, which cannot be obtained with a single molecular configuration. Moreover, the full width at half maximum fitting leads to the difference in the vibration mode of the two configuration results at a measured peak position.
氨基酸是生命蛋白质的基本组成单位, 自然界中发现的氨基酸有三百多种, 被广泛应用于解毒, 转运和物质储存等多个方面, 涉及医药, 食品和化妆品等多个领域。 氨基酸的检测有荧光分光光度法[1], 柱前衍生[2]和柱前衍生[3]的高效液相色谱法, 对检测环境和样品制作要求较高。 生物分子和超导材料等多种物质的低频共振特性位于太赫兹频段, 使得太赫兹时域光谱(THz-TDS)技术表征生物分子振动模式的应用前景广阔; THz-TDS优点有: (1)只需对样品信号进行傅里叶变化即可获得振幅, 相位等信息; (2)采用相干源和高灵敏探测器, 信噪比与动态范围得到提高; (3)样品压片处理和无水检测环境即可。
以亮氨酸和异亮氨酸作为研究对象, 二者同属支链氨基酸, 且化学式一致。 首先, 已有大量文献对其太赫兹(THz)光谱报道[4, 5, 6], 数据详实, 其次, 报道中模拟计算只有单分子构型, 且只使用一种方法[5]。 单分子构型主要针对孤立分子的分子内振动, 对于分子间相互作用较强的物质, 需要考虑周期性结构。 分子振动模式不能认为是局域化的原子振动, 需要从分子整体出发, 包括分子内振动和分子间振动。 此外, 模拟结果主要受理论方法影响, 适合结构计算的方法能够更加精确指认振动模式, 因此, 对研究对象进行单分子和晶胞构型模拟计算, 单分子模拟使用从头算法、 半经验法和密度泛函理论计算, 晶胞模拟使用四种广义梯度近似密度泛函计算, 并指出产生差异的原因, 根据不同构型的多种模拟结果, 在分析以氨基酸为单元的大分子结构时, 能够提供丰富的理论基础。
随着太赫兹科学及应用技术的高速发展, 国内外研究者对氨基酸的太赫兹光谱分析取得突破性进展, 其光谱数据被相继报道, 填补了远红外区的光谱数据空白, 为建立氨基酸的光谱数据库及物质识别奠定基础, 也为相应的理论技术提供可靠的实验数据。
2003年Taday[7]等测量谷氨酸在0.1~3.0 THz波段的吸收谱, 随后Yamaguchi[8]等测量酪氨酸, 天冬氨酸和苯丙氨酸等氨基酸太赫兹吸收谱; 2007年Nishizawa[9]等使用铬镁橄榄石激光器的THz-TDS系统测量20种基本氨基酸太赫兹波段的吸收谱, 2009年王卫宁[4]等测量基本氨基酸的多晶粉末状样品0.2~3.0 THz波段吸收谱, 丰富了氨基酸的光谱数据。 后期, 大量科研机构加入该项研究, 通过探究样品浓度影响及改进探测技术, 进一步提高了氨基酸的太赫兹指纹谱的可靠性。
如表1所示, 列举了前人研究亮氨酸与异亮氨酸位于0.2~2.6 THz波段指纹数据库。 各个研究团队使用不同方法进行检测, 导致吸收峰位及个数有所差异, 文献[5]的实测峰位包含其他文献描述的全部峰位, 因此, 使用该数据作为理论计算的对照。
| 表1 亮氨酸与异亮氨酸指纹数据库 Table 1 Fingerprints database of leucine and isoleucine |
适用于宏观现象的经典力学无法对微观系统中原子与电子的行为正确描述。 1925年, 海森堡(Heisenberg)以矩阵力学的形式创造量子力学, 1926年薛定谔(Schrö dinger)从波动力学角度出发, 提出薛定谔方程[10, 11, 12], 并证明与矩阵力学等价。 这两个理论为人们理解微观世界提供理论依据, 若给定初始和边界条件, 即可求解出波函数, 得到体系的性质。 多粒子体系的定态薛定谔方程为
式(1)中, 等式左侧依次为: (1)原子核动能, (2)电子动能, (3)原子核与电子相互作用势能即吸引能, (4)电子与电子间排斥能, (5)原子核与原子核间排斥能。 实际计算过程中, 大部分原子属于多电子结构, 原子核与电子运动复杂, 受数学计算限制, 仅有简单体系(氢原子, 类氢离子和简谐振子等)能得到精确结果, 多电子体系需要引入若干近似求解, 量子化学计算转变为求解薛定谔方程近似解。
近似(1): 非相对论近似, 研究体系含有限个原子核和电子, 运动速度远小于光速, 不存在产生和湮灭现象, 可以忽略相对论效应。 采用非相对论近似, 薛定谔方程中包含原子核与电子两项, 难以求解, 因此引入近似(2)和(3): 玻恩-奥本海默(Born-Oppenheimer)近似[13], 原子核的质量约为电子质量的104倍, 原子核的运动速度远小于电子, 当原子核进行微小运动, 电子都迅速运动调整, 形成与原子核力场对应状态。 玻恩和奥本海默基于上述思想, 将分子中原子核的运动与电子运动分开考虑, 即计算原子核运动时不考虑电子空间分布变化, 认为快速运动的电子建立平均化的电子分布, 计算电子运动时认为原子核固定不动; 单电子近似, 基于玻恩-奥本海默近似, 原子核固定时体系的电子运动方程为
式(2)中, rij涉及两个电子坐标, 无法进行分离变量, 因此近似为每个电子均独立运动于核库伦场与其他电子的平均势叠加的势场, 单电子运动特性只取决于其他电子密度分布, 不考虑电子间相互作用。 用单电子波函数之积描述体系总电子波函数, 从而将电子间势能能变成与rij无关的函数, 便于求解。
常用的量子化学计算方法有分子轨道从头计算法(ab inito), 半经验分子轨道法(semiempirical)和密度泛函理论(density functional theory, DFT)等。 其中, 从头计算法求解薛定谔方程过程中, 除上述三个基本近似外, 不再引用其他近似, 仅使用基本的物理常数(普朗克常数, 光速等)作为已知参数, 不引入任何经验参数, 完全利用数学工具严格计算各类积分, 它是非经验的, 全电子的方法; 半经验分子轨道方法求解薛定谔方程, 并非使用原始的, 完整的哈密顿算符, 而是包含一些待定参数, 用参量化办法估计方程中影响较大的积分值, 例如, 用光谱数据估计, 或用参量函数计算。 此外, 半经验方法全部或部分省略多中心积分; 密度泛函理论认为体系的基态能量仅仅是电子密度的泛函, 以及以基态密度为变量, 将体系能量最小化之后就得到了基态能量; 目前其发展主要针对交换关联泛函展开, 例如, 广义梯度近似利用梯度展开交换相关能, 使其既是电子密度泛函, 又是电子密度梯度泛函。 广义梯度近似交换相关能量为
式(3)中, ∇ρ 为电子密度梯度。 PBE[14]泛函为更好描述广义梯度近似的非局域域性, 对式(3)加入交换能量增强因子FX(S), 定义为
式(4)中, κ =0.804, μ =0.219 51。 PBEsol泛函[15]认为获得原子准确交换能量, 需要强烈违反缓慢变化的密度梯度, 并且恢复梯度扩展应该改进描述, 增强因子为
保留PBE泛函的其他条件; RPBE泛函[16]增强因子为
式(6)中, κ =0.804; WC泛函[17]认为增强因子取决于过程的近似值和对参数的拟合, 因此只能得到定性特征, 需要选择已知约束, 将式(5)的μ s2设为x,
式(7)中, c=0.007 932 5。
杂化泛函引入部分Hartree-Fock方法计算的交换能, 并与密度泛函理论计算结果线性结合, 获得更精确的交换关联能。 例如, B3LYP泛函[18]表达式为
式(8)中, a0=0.20, aχ =0.72, aC=0.81; M06-2X泛函[19]表达式为
式(9)中, X=54, 与B3LYP泛函相比,
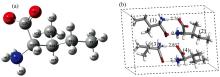
计算的输入构型来自剑桥晶体数据库(CCDC), 模型在室温(283~303 K)条件利用中子衍射技术获得并修正, 亮氨酸CCDC编号为1206031, 异亮氨酸CCDC编号为1542778。 图1(a)为α -亮氨酸(C6H13NO2)单分子构型, 存在COO-和
 | 图1 亮氨酸构型图 (a): 单分子; (b): 晶胞Fig.1 Structure of leucine (a): Single molecule; (b): Unit cell |
 | 图2 异亮氨酸构型图 (a): 单分子; (b): 晶胞Fig.2 Structure of isoleucine (a): Single molecule; (b): Unit cell |
单分子模拟利用Gaussian09软件进行结构优化和振动分析, 使用半经验法(PM6), 从头计算法(HF, MP2)和密度泛函理论(B3LYP, M06-2X)共五种方法结合6-311+G(d, p)高斯型基组, 其中, 除HF方法外, 均加入色散修正(D3)。 分子振动基于谐振计算, 考虑实验存在非谐振效应, 故计算结果使用标度因子修正。
晶胞模拟利用Materials Studio 2019软件进行计算, 该软件考虑固体的周期晶格排布。 使用广义梯度近似(GGA)的PBE, PBEsol, RPBE和WC共四种密度泛函, 采用拟牛顿算法的线性搜索(BFGS)和模守恒赝势(Norm-conserved), 截断能设置为830 eV。 避免优化后晶胞参数变化导致峰位偏差过大, 因此, 计算时固定晶胞的几何形状, 只优化原子位置。
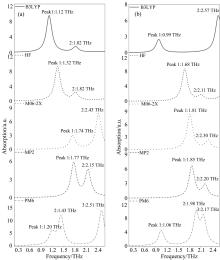
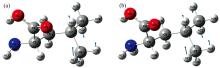
如图3所示, PM6方法对亮氨酸和异亮氨酸模拟计算得到三个峰位, 其余方法均得到两个峰位, 说明半经验法引入大量参数, 虽然能减少计算资源, 但计算结果的准确性还需进一步探究。 该模拟计算的振动频率必然是分子内振动, 与实验数据对比, 吸收峰值位置和强度均有很大偏差, 并且不能仅以一种方法的峰位作为振动模式指认, 如图4所示, B3LYP方法1.12 THz处振动模式与HF方法1.32 THz处的振动模式相同, 为C8-C7-C9扭转, 若只匹配峰位, 可分别对应实测0.85, 1.46的两个峰位, 存在一定局限性。 除未考虑分子间振动外, 还有Gaussian软件以分子轨道为基础, 认为电子轨道不是各原子的定域, 因此结构优化结果为能量更低的分子形式, 对模拟计算产生一定影响。 单分子构型计算意义(1)为含量较低的样品THz吸收谱提供多种理论数据参考, (2)对由多种分子组成的样品的成份分析。
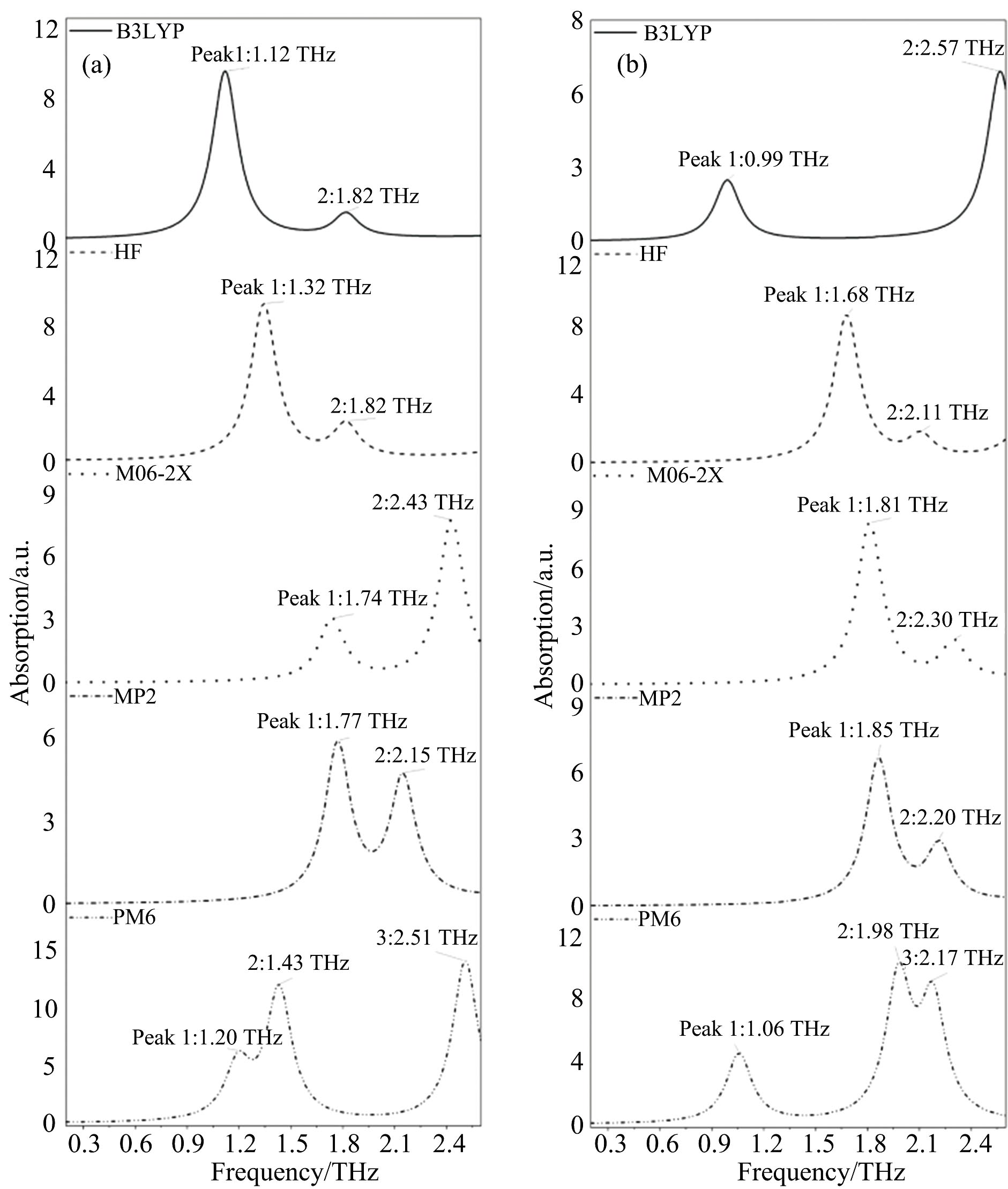 | 图3 单分子构型模拟吸收谱图 (a): 亮氨酸; (b): 异亮氨酸Fig.3 Simulated absorption spectra of single molecule structure (a): Leucine; (b): Isoleucine |
 | 图4 单分子构型振动模式 (a): B3LYP-1.12 THz; (b): HF-1.32 THzFig.4 Single molecule structure vibrational modes (a): B3LYP-1.12 THz; (b): HF-1.32 THz |
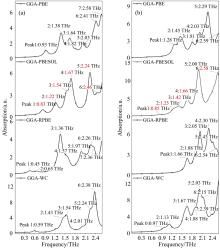
如图5所示, 晶胞构型模拟结果在吸收峰位和个数与实测值匹配度得到显著提升, 但不同密度泛函结果存在一定差异, 其中, PBE, RPBE和WC三种泛函只对引入的经验参数改变, PBEsol泛函通过改变交换关联能梯度展开式的二阶系数改进了对固体交换能的描述, 并且量子化学计算都存在近似误差, 还受到迭代收敛标准影响, 因此与实测结果均存在一定红移或蓝移。 如表2和表3所示, 为优化后重原子结构参数, 整体来看, 亮氨酸和异亮氨酸使用四种泛函优化后结构接近, 说明结构优化对泛函选择要求不高, 但其他性质计算需要考虑泛函与结构的适用性。 二者使用PBEsol泛函模拟吸收峰位与实测值最接近, 但结构参数及分子位置与输入结构差异并不最小, 说明不能仅以结构参数变化判断泛函是否适用计算, 可能由于输入构型存在一定误差。 此外, 验证了PBEsol泛函获得原子能量更准确, 且对异构体模拟计算选择相同泛函计算即可, 减少测试计算资源。
 | 图5 晶胞构型模拟吸收谱图 (a): 亮氨酸; (b): 异亮氨酸Fig.5 Simulated absorption spectra of unit cell structure (a): Leucine; (b): Isoleucine |
| 表2 亮氨酸晶胞构型几何计算 Table 2 Geometric calculation of Leucine unit cell structure |
| 表3 异亮氨酸晶胞构型几何计算 Table 3 Geometric calculation of unit cell structure of isoleucine |
使用PBEsol泛函模拟结果对应亮氨酸与异亮氨酸的振动模式指认, 利用Materials Studio软件振动演示功能, 将其指认于表4, 其中, 亮氨酸2.24与1.67 THz振动模式相比, #2、 #4振动方向相反; 异亮氨酸2.58与0.85 THz振动模式相比, 除振动方向不同外, 2.58 THz处COO-表现为扭转振动。
| 表4 实测和理论计算吸收峰位及相应频谱标定 Table 4 Absorption peaks of experiment and simulated, and related vibration rotation modes |
振动模式指认结果表明, 振动模式除分子间相互作用外, 还有部分来源于分子内振动, 这一部分无论是峰位或振动模式, 与单分子模拟结果并不一致, 产生原因为晶胞模拟计算得到大量振动模式, 利用洛伦兹模型对其进行半峰全宽(full width at half maximum, FWHM)得到模拟谱, 一些强度较低或拟合为非峰位的振动并不会表现出来。 实验中未测得峰位的原因有: (1)实验温度为室温, 模拟计算温度为0 K; Shen[20]等验证低温情况会发现一些新的吸收峰, 且随温度升高强度降低; (2)密度泛函高估该模式的振动强度; (3)测量仪器分辨率不够, 超出检测范围或无法区分频率接近的振动。
研究了亮氨酸与异亮氨酸在0.2~2.6 THz波段的特征指纹峰, 并根据PBEsol泛函模拟结果精确指认振动模式。 通过不同构型的模拟计算, 说明单分子构型模拟计算存在一定局限性; 晶胞构型模拟计算需要选择合适的密度泛函才能揭示吸收谱形成机理。 异构体虽然部分实测吸收峰接近, 但振动模式差异很大, 无论实测还是理论计算, 均可对其进行区分, 且异构体的理论计算选择同一泛函即可, 能够节约部分计算资源。 研究结果为识别亮氨酸与异亮氨酸提供丰富理论依据, 为多糖、 蛋白质等生物大分子量子化学计算提供理论参考。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|