

{kind=link}
{kind=link}
{kind=link}
电子圆二色光谱图的模拟与软件开发
[许范2  , 苏俊奎
, 苏俊奎1 , 文燕1 , 甘礼社1, 2, * ]
, 苏俊奎]
|
|


作者简介: 许 范, 女, 1997年生, 浙江大学药学院现代中药研究所硕士研究生 e-mail: 21919057@zju.edu.cn
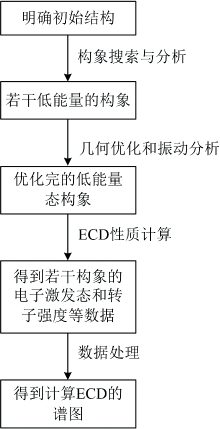
有机小分子化合物, 尤其是天然产物具有复杂和多样的结构, 且一般含有多个手性中心, 确定其绝对构型一直是结构鉴定的难点之一。 电子圆二色光谱(ECD)具有样品无损、 曲线简洁等优势, 是鉴定绝对构型的强有力的手段之一。 利用密度泛函理论(DFT)和含时密度泛函理论(TDDFT)等量子化学理论以及计算机技术, 对化合物的理论ECD计算模拟, 与实验值对比确定绝对构型是目前主流技术。 计算模拟流程如下: 首先确定化合物的相对构型, 其次运用构象搜索软件对化合物进行构象分析, 然后对搜索得到的多个最低能量态构象(如能量阈值在2.0 kcal·mol-1以内)进行几何优化和振动分析, 将已经优化完成的优势构象投入ECD计算, 计算ECD相关参数, 得到电子激发能(eV)和转子强度(10-40 erg-esu-cm)等数据, 最终根据数据进行多构象谱图拟合。 该研究主要对ECD曲线的模拟方法、 ECD计算结果的评价标准进行了全面总结。 在此基础上, 运用Python语言开发了一款自动化处理高斯ECD计算结果、 模拟ECD曲线的软件, 该软件能够对计算得到的输出文件进行定位, 对于化合物几何优化和振动分析得到的输出文件, 可以抓取各个构象的吉布斯自由能数据, 并且依据此数据进行玻尔兹曼分布的加权, 对于计算ECD相关性质的输出文件, 可以抓取ECD计算相关的电子激发能和转子强度的数据, 依据以上数据得到的ECD计算理论光谱图, 能够进行实时模拟参数调节并与实测曲线对比, 方便了计算ECD的数据处理和结果评价。
Organic small molecules, especially natural products, have complex and diverse structures and generally contain multiple chiral centers, so the determination of their absolute configuration has always been a challenge in structural elucidation. Electronic circular dichroism (ECD) is a powerful tool for identifying absolute configuration because of its concise curve and without sample loss. With effective use of computer technology and quantum chemistry theories such as density functional theory (DFT) and time-dependent density functional theory (TDDFT), the calculation of theoretical ECD spectrum and comparison with the experimental one has become a state-of-the-art technique for absolute configuration determination. The main process is as follows: First, the relative configuration of the compound needs to be determined before calculation. Conformation analyses are carried out to identify multiple lowest energy conformers, which are then subjected to geometric optimization and vibration analysis (such as the energy threshold within 2.0 kcal·mol-1). ECD-related parameters, such as electronic excitation energy (eV) and rotor strength (10-40 erg-esu-cm), are further calculated for each lowest energy conformer. Finally, these data are processed according to Gaussian broadening function for spectrum fitting, and the calculated spectrum of all conformers is averaged according to their Boltzman weight. This article mainly summarizes the simulation method of the ECD curve and the evaluation standards of the ECD calculation result. Furthermore, software was developed for automatically processing Gaussian ECD calculation results and simulation of ECD curve based on Python language. The software can locate keywords in the output files. For the geometry optimization and vibration analysis output files, it can capture the Gibbs free energy data of each conformer and weight the Boltzmann distribution. The ECD-related properties output files can capture the excitation energy (eV), the corresponding oscillator and rotatory strength, and simulate the theoretical spectrum. The software can adjust simulation parameters in real-time and compare them with the experimental curve, facilitating the data processing and result evaluation of the calculated ECD.
有机小分子化合物尤其是天然产物, 一般具有多个手性中心, 而确定手性中心的绝对构型是结构鉴定中的难点之一。 目前确定手性分子绝对构型的方法有: 手性有机合成; 基于NMR的Mosher法或位移试剂法[1]; X射线单晶衍射; 手性光谱学方法等。 在这些方法中, 相对于手性有机合成过程的繁琐复杂; 基于NMR的Mosher法中要求化合物中必须含有易于衍生化的基团、 需要昂贵的手性试剂而且会对样品造成损失; 以及基于Bijvoet法的X射线单晶衍射(或Cu靶)需要样品纯度高、 合适的培养条件等要求; 手性光谱学方法对于样品的纯度、 能够结晶等条件要求不高, 测量所需样品量少, 测量之后样品可回收, 因此得到了广泛地应用。
电子圆二色光谱(electronic circular dichroism, ECD)是手性光谱法中最常用的手段之一。 通过手性化合物溶液的左旋圆偏光和右旋圆偏光的吸收系数之差Δ ε , Δ ε 随波长变化即可获得圆二色光谱, 它对绝对构型和构象特征十分敏感[2]。 传统的圆二色光谱所用的平面偏振光的波长范围一般在200~400 nm, 属于紫外区, 由于其吸收光谱是分子电子能级跃迁引起的, 称为电子圆二色光谱。
目前, 传统的半经验规律已经难以满足对ECD谱图的解释, 随着计算机技术的迅速发展以及密度泛函理论(density functional theory, DFT)[3]和含时密度泛函理论(time-dependent density functional theory, TDDFT)[4, 5]的成熟, 量子化学计算已经成为一种越来越强大的工具。 通过ECD的理论计算与实验值进行比较, 已成为确定有机小分子绝对构型的有效流行技术[6, 7, 8]。
在ECD计算模拟过程中, 常常涉及多个构象的数据处理、 加合、 参数调整、 评价等繁琐过程, 导致数据处理量大, 标准不统一。 本课题组一直致力于ECD的计算模拟确定小分子绝对构型的研究, 在本文中, 我们对目前ECD模拟技术方法进行了总结, 更利用Python语言开发了一款自动化处理高斯ECD计算结果、 模拟ECD曲线的软件(ECD Curve, Windows版), 能够进行实时模拟参数调节并与实测曲线对比。 该软件专门针对ECD的Gaussian计算结果, 依据高斯函数展宽, 从而得到某个构象的理论光谱图, 只需将输出文件拖拽进入相应的文件框中, 即可自动生成相应的理论光谱图, 对半高全宽等参数的调整即可获得该化合物的理论光谱图, 极大地方便了ECD计算结果的数据处理和作图过程。
计算ECD常用的量子化学程序有Gaussian[9]和Dalton[10]两种软件, 其中Dalton是免费软件, 主要用来计算闭壳层的基态。 Gaussian是一款商用软件, 可在不同型号的大型计算机, 超级计算机, 工作站和个人计算机上运行, 具备Linux版本和Windows版本, 并且搭配GaussView的可视化软件, 是目前计算化学领域内流行、 应用范围广的综合性量子化学计算程序包。
1.2.1 ECD理论光谱模拟原理
计算所得的圆二色光谱数据主要是化合物的某一构象在一系列能量状态(波长)下的电子激发能(eV)和转子强度的数据(10-40 erg-esu-cm), 由于在实验中得到的圆二色光谱的吸收带的强度是在一段波长范围内分布的, 因此需要用到Gaussian函数进行吸收带的线性模拟展宽, 其理论公式表示如式(1)
式(1)中, Δ Ei和Ri分别代表i态下的电子激发能和转子强度, σ 为谱峰的半高全宽, 这些均是调整ECD理论光谱图的重要参数。
1.2.2 玻尔兹曼分布理论
一个化合物在溶液中是由多种构象组成的一个混合体, 而每个构象根据其能量的大小占全部构象的比例服从玻尔兹曼分布
式(2)中, ρ i为i构象所占比例, i、 j为构象编号; ni为处于第i构象的分子数; Δ Gi是指构象i相对于最低能构象的吉布斯自由能(kcal· mol-1); T为温度, 一般为室温(298.15 K); R理想气体常数(0.001 985 899 5)。
1.3.1 数据处理和谱图拟合
(1)利用Excel数组公式实现的数据处理方法
数组公式是公式在以数组为参数时的一种应用。 数组公式可以看成是有多重数值的公式。 与单值公式的不同之处在于它可以产生一个以上的结果。 本课题组利用Excel数组公式实现计算ECD的理论光图谱的模拟, 主要根据前述1.2.1介绍的ECD计算的高斯函数展宽公式, 将ECD计算得到的第1~60个电子激发能(eV)和转子强度的数据(10-40 erg-esu-cm)分别填入Excel中。 其中A列A1:A60为该构象计算所得的60个激发态的电子激发能, B列B1:B60为对应的60个激发态的转子强度。 在A列61中设置一个波长(如150~450 nm中的起始150 nm), 则B列61中可用数组公式=SUM((A$1:A$60)* (B$1:B$60)* EXP(-POWER(((1240/A61)-(A$1:A$60))/($C$1), 2)))/(22.96* ($C$1)* SQRT(PI()))计算出该波长下的理论ECD吸收值(Δ ε )。 以此类推, 填满150~450 nm波长范围, 就可以自动求出每个波长下的数值, 从而获得某个构象的ECD曲线数据。 多个优势构象又依据玻尔兹曼分布占比在Excel表格中设定公式进行加权, 生成该化合物的总的计算ECD的理论光谱图。 将此编辑完成的Excel表格保存, 在Origin软件中打开, 以两列数据(X轴为波长、 Y轴为Δ ε )作图, 可以得到最终呈现的ECD理论光谱图。
 | 图1 ECD计算的流程图Fig.1 Flow chart of ECD calculation |
(2)运用已开发的软件实现的方法
除了Gaussview软件之外, SpecDis[11]是一个处理计算化学结果的小工具。 此工具需要先提取几何优化和振动分析后得到的输出文件中的吉布斯自由能, 并且自动新建gibb的文件夹, 将所有构象的吉布斯自由能保存成为allheats.txt之后, 抓取与计算ECD相关的激发态等参数, 自动新建spectra的文件夹, 生成相应的各个构象的* .bil文件。 最后, 需要在此选择路径打开之前的gibb的文件夹和各个构象的* .bil文件, 相应的计算ECD曲线生成。 就整个操作流程来说, 操作步骤过多, 略显繁琐, 对于数据处理过程不够简便。
Multiwfn[12]是由卢天开发的一款功能强大的波函数分析软件, 但也需要将优化完生成的吉布斯自由能以及各个构象相应的玻尔兹曼分布占比需要汇总在命名为multiple.txt的文件中, 并且与相应的计算ECD相关参数的输出文件保存在同一路径下。 整体来说, 流程不够简便, 一旦涉及多构象的处理就显得繁琐。
1.3.2 理论ECD和实测ECD对比
在ECD光谱图中呈现正峰的为正cotton效应, 负峰的为负cotton效应。 最终确定化合物绝对构型主要是将计算得到的ECD的理论光谱图与通过圆二色光谱仪检测得到的实验ECD光谱图进行对比, 观察两者出现的cotton效应与波长是否一致。 当然, ECD理论光谱图可以进行适当的调整, 使得ECD的理论光谱图与实测的ECD光谱图更好地吻合, 但也应该限定在有限的范围内, 否则极容易出错。 一般设置在0.2~0.6 eV范围内, 以0.4为最常见。 由于实际测定过程中能量的衰减和耗散, 预测的理论ECD 光谱相对实测值可能会有一定的整体偏移, 通常可以进行在-30~+30 nm之间的平移[13, 14]。 除了肉眼对比观察之外, SpecDis引入相似因子的概念以量化两种曲线之间的匹配程度, 这是一种发展趋势[11]。
激发态的计算不能提供完整的曲线, 而只能给出每个跃迁的激发态相对应的转子强度, 而要将这些数据转化为ECD的理论光谱, 需要使用前述提及的Gaussian函数等进行理论展宽。 本软件主要运用Python语言编写代码, 针对几何优化和振动分析的计算环节中得到的输出文件进行数据定位, 抓取得到各个构象的吉布斯自由能, 并且根据前述提及的玻尔兹曼分布理论进行处理, 得到各个构象的玻尔兹曼分布占比。 同时, 将ECD性质计算得到的输出文件进行数据定位, 抓取得到与ECD计算相关的电子激发能(eV)和转子强度的数据(10-40 erg-esu-cm), 依据Gaussian函数展宽, 得到ECD计算的理论光谱图。 基于以上理论及数据处理, 运用Python语言, 从而开发得到一款可以调节半高全宽σ 、 构象个数以及波长范围的自动化处理Gaussian ECD计算结果的软件。
对比目前已开发的软件和利用Excel数组公式实现的数据处理方法, 本课题组开发的软件操作流程更加简单, 只需直接将几何优化和振动分析的输出文件和计算ECD性质的输出文件拖拽进入相应页面板块, 即可生成相应的计算曲线, ECD计算谱图的数据处理的操作流程变得更加便捷。
该软件图形界面主要由谱图生成板块、 优化生成的输出文件板块、 ECD生成的输出文件板块以及参数设置板块这四个板块组成。 将优化生成的各个优势构象的输出文件直接拖拽至OPT板块, 各个构象会依据玻尔兹曼分布占比显示在Weight列中, 然后将ECD计算生成的输出文件拖拽至ECD板块, 然后将parm中填写σ 为半高全宽的数值, X1为波长λ 的起始波长, X2为波长λ 的结束波长。
 | 图2 软件ECD Curve的界面Fig.2 Interface of ECD curve |
将σ 、 X1和X2进行修改, 或者单独点击log-fit、 curve-fit或者all-fit, 点击generate, 可以生成相应的修改之后的曲线。
该软件免费供测试使用, 网址链接如下: https://www.onlinedown.net/soft/10019240.htm。
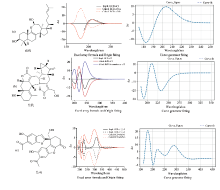
选取三个化合物(见图3), 分别用Excel数组公式拟合和使用软件ECD Curve进行拟合, 两相对比, 验证该软件的吻合程度。
根据以上三个化合物的ECD理论光谱的拟合对比来看, 由于Origin处理Excel表格数据时, 会删减激发态以更好得拟合实测的ECD结果, 所以拟合的差异主要在200 nm以下。 从整体来看, 用软件ECD Curve能很好地重现数组公式的结果, 从而证明可以用该软件处理ECD的数据获得可靠的模拟结果。
ECD是最成熟的手性光学方法之一, 在过去的几十年中经历了巨大的技术发展, ECD无疑仍然是解决各种立体化学和分析问题的最广泛使用的手性技术之一。 本研究对ECD曲线的模拟方法、 ECD计算结果的评价标准进行了总结, 在此基础上, 运用Python语言开发了一款自动化处理高斯ECD计算结果、 模拟ECD曲线的软件, 能够进行实时模拟参数调节并与实测曲线对比, 该软件使用方便, 省去了使用Excel数组公式拟合曲线的繁琐步骤, 且对比之下拟合的理论光谱图并无明显差异, 这极大缩短了我们在通过计算确定化合物绝对构型过程中数据处理的过程, 很大程度上方便了我们对于ECD的理论光谱图的获取及其与实测值对比中的实时参数调整。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|