
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
C15NO4H nI m ( n=11, 12, 13, m=4, 3, 2, n+m=15)结构与光谱性质的密度泛函理论研究
[郭雅晶1  , 李秀燕
, 李秀燕2 ]
, 李秀燕|
|


作者简介: 郭雅晶, 女, 1986年生, 太原师范学院物理系高级实验师 e-mail: guoyajing58@163.com
二碘甲状腺原氨酸(分子式为C15NO4H13I2), 三碘甲状腺原氨酸(分子式为C15NO4H12I3)以及四碘甲状腺原氨酸(分子式为C15NO4H11I4)作为甲状腺分泌的主要激素, 对人脑的发育、 神经递质的合成、 新陈代谢的调节和甲状腺功能正常运作等起到至关重要的作用。 通过理论计算方式对C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15)三种化合物进行系统研究, 为今后的实验研究提供详实的理论依据。 通过Gaussian软件包和GaussView软件结合进行理论计算, 在选用B3LYP/Lanl2mb基组水平上, 首先采用密度泛函理论(DFT)优化了C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15)三种团簇的几何和电子结构。 然后基于这些团簇的基态稳定结构下, 其激发态吸收谱和发射谱的研究使用相同的基组水平并采用极化连续介质模型(PCM)下运用含时密度泛函理论(TDDFT)进行。 研究结果表明, 优化所得C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15)团簇的几何结构对称性均为C1; 在C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15)基态稳定结构基础上, 得出它们的输运性质, 即二碘甲状腺分子(C15NO4H13I2)既不具有p型输运性质亦不具有n型输运性质, 而三碘甲状腺素(C15NO4H12I3)和四碘甲状腺素(C15NO4H11I4)团簇均为p型输运材料; 通过含时密度泛函理论, 在优化好的基态结构基础上, 又计算了它的溶剂效应, 进一步得出该分子在水溶剂中的吸收谱特性, 同时通过研究还获得了C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15)团簇的手性光谱ECD特征图谱。 理论研究可以为实验研究提供可对比的理论数值, 为今后的实验研究提供了可行性参照值。
Diiodothyronine (formula C15NO4H13I2), triiodothyronine (formula C15NO4H12I3), and tetraiodothyronine (formula C15NO4H11I4) are the major hormones secreted by the thyroid gland, which contribute to the development of the human brain, the synthesis of neurotransmitters, the regulation of metabolism and the normal functioning of thyroid function playing a crucial role. Three compounds of C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15) have been studied systematically by theoretical calculations in this paper, which provides a detailed theoretical basis for future experimental research. In this research, combining the Gaussian software package and GaussView software to carry out theoretical calculation, the geometrical and electronic structures of C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15) clusters are optimized by using density functional theory (DFT) at the B3LYP/Lanl2mb level. Then, based on these clusters' stable ground state structure, the excited state absorption and emission spectra are studied at the same basis set level using the polarized continuum model (PCM) with time-dependent density functional theory (TDDFT). The results show that the geometrical structure symmetry of the optimized C15NO4H nI m ( n=11, 12, 13, m=4, 3, 2, n+ m=15) clustersare C1; based on the stable structure of the ground state for C15NO4H nI m ( n=11, 12, 13, m=4, 3, 2, n+ m=15) clusters, the transport properties are obtained, C15NO4H13I2has neither p-type transport property nor n-type transport property, the C15NO4H12I3 and C15NO4H11I4 clustersare p-type transport material; and then, based on the theory of time-dependent density functional, the solvent effect is calculated based on the optimized ground state structures. Meanwhile, the absorption spectra characteristics of the molecules in water solventare further obtained, and the chiral spectra of the C15NO4H nI m( n=11, 12, 13, m=4, 3, 2, n+ m=15) clusters are also studied by ECD. The theoretical research can provide a comparable theoretical value for the experimental research and a feasible reference value for future experimental research.
二碘甲状腺原氨酸(又名二碘甲状腺素, 英文简写T2)是甲状腺分泌的主要激素之一, 同样包括三碘甲状腺原氨酸(又名三碘甲状腺素, 英文缩写为T3), 四碘甲状腺素(又名四碘甲状腺原氨酸, 英文缩写T4)也是甲状腺分泌的主要激素, 其分子式分别为C15NO4H13I2、 C15NO4H12I3和C15NO4H11I4。 二碘甲状腺激素和四碘甲状腺素对人脑的发育和成熟至关重要, 并负责神经递质合成所需关键酶的合成[1, 2, 3]; 三碘甲状腺素测定可用于T3-甲亢的诊断, 早期甲亢的查明和假性甲状腺毒症的诊断[4]; 四碘甲状腺激素由甲状腺在碘存在下产生, 负责调节代谢, 碘的缺乏会导致四碘甲状腺素的产生减少, 并扩大甲状腺组织和引起甲状腺肿大[5, 6, 7]。 Petruk等的3, 5-二碘酰胸腺苷的红外光谱和拉曼光谱比较分析表明, 3, 5-二碘基胸腺苷和色氨酸在晶体状态下芳香环振动产生四种中强拉曼, 频率在1 530和1 620 cm-1之间[8]。 除此以外, Fortino研究组针对四碘甲状腺素(T4)具有运载转移酶和蛋白的作用进行了生物实验研究[9]。 Alvarez等研究了三碘甲状腺素分子的合成过程, 及生物药理性质的实验研究[10]。 Alvarez研究组记录了四碘甲状腺素的傅里叶变换红外光谱150~4 000 cm-1频率范围内的频谱和拉曼频谱[11]。
诸多学者均从实验角度对二碘甲状腺素, 三碘甲状腺原氨酸, 四碘甲状腺素进行了生物化学, 药物医理和药物合成等研究, 但未从理论计算角度对这些物质进行物理化学性质, 光谱性质和电荷性质等方面分析研究, 因此, 有必要对二碘甲状腺素, 三碘甲状腺原氨酸, 四碘甲状腺素进行详细的理论研究, 以了解三种分子的性质和功能; 有必要对这三种生物分子进行详细的光谱研究, 以深入了解其结构、 输运特征、 分子振动特性和光谱分布特性。 理论研究可以预测实验结果, 同时可以和实验结论进行对比, 有时理论结果与实验结果会有细微的偏差, 这是由于理论计算环境单纯, 无外界干扰, 实验过程甚至结果会受外部环境的影响, 因此会出现细微偏差; 在选取点群结构合理以及优化泛函和基组水平合理的基础上, 理论与实验之间的相对误差可以低于1%[12], 所以合理的理论计算可满足多数实际应用的需要。
为了进一步研究二碘甲状腺素, 三碘甲状腺原氨酸, 四碘甲状腺素的几何电子结构、 微观物理化学性质、 微观内部电子输运性质、 芳香性、 红外与拉曼光谱以及其在水溶剂作用下的吸收和发射光谱性质, 本工作采用密度泛函理论和含时密度泛函理论在B3LYP/Lanl2mb基组水平上对二碘甲状腺素(T2)、 三碘甲状腺原氨酸(T3)、 四碘甲状腺素(T4)进行理论研究。
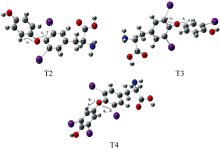
所有计算均使用Gaussian 09软件包进行[13], 采用Becker的三参数混合泛函结合Lee, Yang和Parr相关泛函(B3LYP)[14]并在Lanl2mb基组水平下, 运用密度泛函理论(DFT)研究了三种甲状腺素分子(T2、 T3、 T4)的结构。 T2, T3, T4分子几何结构图像(如图1)通过GaussView5.0软件生成[15], 预测了甲状腺素团簇的不同自旋多重性和初始结构; 为了证实甲状腺素团簇结构的稳定性, 分析了该分子的简谐振动频率; 由此得到能量最低且无虚频(NIMAG=0)的甲状腺素团簇的基态稳定结构。 在该团簇的基态稳定结构下, 研究了它的电离势(IP)、 电子亲和能(EA)、 提取能(HEP/EEP)以及重组能(λ h/λ e), 用以表征该团簇的输运特征; 此外, 研究了这些分子的芳香性和红外(IR)以及拉曼(Raman)分子振动谱。 最后, 选择极化连续介质模型(PCM)中的自洽反应场(SCRF)计算T2、 T3、 T4分子化合物的溶剂效应, 具体理论计算方法是在相同基组(B3LYP/Lanl2mb)水平上采用含时密度泛函理论(TDDFT)进行了吸收光谱和发射光谱的研究, 并采用极化连续介质模型(PCM)在H2O溶剂中求解nstates=20的激发态光谱。
 | 图1 T2, T3, T4团簇几何结构Fig.1 Structures of T2, T3, T4 clusters |
四碘甲状腺素(T4)团簇的分子式为C15H11I4NO4, 三碘甲状腺素(T3)团簇的分子式为C15H12I3NO4, 二碘甲状腺素(T2)团簇的分子式为C15H13I2NO4, 经过B3LYP/Lanl2mb优化以及计算频率, 确定了最低能量稳定结构(如图1所示)。 图1中灰色原子为碳原子(C), 白色原子为氢原子(H), 红色原子为氧原子(O), 紫色原子为碘原子(I), 蓝色原子为氮原子(N), 每个原子符号前的数字为设置分子结构时原子排列顺序数。 计算过程中环境参数设置为大气压1.000 Atm, 温度为298.150 Kelvin。
表1列出了优化后所得T2, T3, T4分子内I— C键长和分子内苯基上的C— C与C— O键长, 苯基C— C— C键角和分子内C— O— C键角以及扭转角θ 1和θ 2(扭转角均选在C— O— C键角变形处), 并将所得结果与Camerman[16]等以及Alvarez[10, 11]等的实验结果进行对比可得出, 文中所选取基组水平得到的数据与实验值相对吻合(例如: 对于T4来说, 其C— C, C— O以及C— I之间键长, 理论计算1.40、 1.39、 2.12 Å 与Camerman等实验计算结果1.38、 1.34、 2.11 Å 几乎一致; 文中所选基组水平得到T3的苯基C— C— C键角和分子内C— O— C键角与Arthur等研究小组所得结果基本一致, 分别为120.0° 和122.2° ; T3扭转角θ 1和θ 2所得值与Arthur等的实验结果存在一定偏差, 这是由于理论计算环境较单一, 不受外界杂质以及振动等因素影响, 故所得结果与实验值吻合的基础上又存在一定偏差; T2的苯基C— C— C键角和分子内C— O— C键角与Alvarez等研究小组所得结果基本一致, 分别为120.0° 和121.3° )。 进一步说明, 理论计算所选取的B3LYP/Lanl2mb基组水平和方法是合理的, 优化所得T2, T3, T4结构是正确的。
| 表1 T2, T3, T4主要键长(Å )、 键角(° )和扭转角θ 1、 θ 2(° ) Table 1 The main bond lengths(Å ), bond angles (° ) and torsion angles θ 1, θ 2 (° ) of T2, T3, T4 |
表2给出了B3LYP/Lanl2mb计算所得的基态参数, 即基态对称性、 最低频率、 结合能、 平均结合能、 温度、 压力、 零点能、 最高占据能HOMO、 最低未被占据能LUMO和能隙。 T2, T3, T4分子的点群(基态对称性)参数均为C1, 使用该水平基组计算的最低频率都是正数(分别为7.73、 6.38和8.20 cm-1), 这意味着它们都位于势能面的局部最小值, 即NIMAG=0(无虚频)。 基态结构下T2, T3, T4结合能与平均结合能的计算结果均为负数, 表2中的结构参数进一步说明B3LYP/Lanl2mb下优化后的三种团簇是基态稳定结构。 此外, 能量间隙是指最高占据轨道(HOMO)能量值与最低未被占据轨道(LUMO)能量值之差的绝对值。 结合上述分析可得出, 所选取的理论计算方法及基组水平是合理的。
| 表2 T2, T3, T4分子基态参数 Table 2 The ground state parameters of T2, T3, T4 |
表3给出了T2、 T3、 T4的电离势(IP, eV)、 电子亲和能(EA, eV)、 空穴/电子提取势(HEP/EEP, eV)和空穴/电子重组能量(λ h/λ e, eV)。 一般来讲, p型输运材料的IPA值在5.680~6.786 eV范围内, n型输运材料EAA在2.411~3.141范围内, IPA和EAA材料的双极性传递应在5.905~7.026和2.797~3.479 eV范围内[17]。 载流子的传输与前线分子轨道的分布密切相关, 通常LUMO (HOMO)能级的离域化程度越大, 越有利于电子(空穴)的传输[18]。 从表3可以看出, T2分子既非p型输运材料, 亦非n型输运材料, 也不具有双极性传递性质; T3分子属于p型输运材料; T4分子属于p型输运材料。 知道T2, T3, T4具有何种输运性质后, 就可以分别选择适合这三种团簇输运的酶或蛋白质进行转运, 也为以后的生物制药提供了一定的理论依据。
| 表3 T2, T3, T4团簇的电离势(IP, eV)、 电子亲和能(EA, eV)、 空穴/电子提取势(HEP/EEP, eV)和空穴/电子重组能(λ h/λ e, eV) Table 3 Ionization potentials (IP, eV), electronic affinities (EA, eV), hole/electron extraction potentials (HEP/EEP, eV) and hole/electron reorganization energies (λ h/λ e, eV) of T2, T3, T4 clusters |
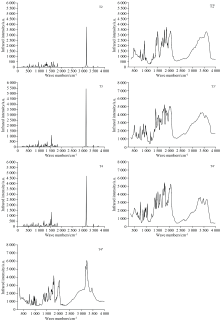
图2给出了T2, T3, T4分子在基态结构下的红外(IR)振动谱, 图中T2, T3, T4图谱为理论计算所得结果, T2', T3', T4', T4″图谱由日本有机化合物数据库SDBS查询到的标准物质红外光谱图[19], 其中T2', T3', T4'图谱通过KBr压片法测得[20], T4″图谱通过石蜡糊法测得。 计算结果与实验结果对比如图2所示, 计算所得IR光谱分布范围与实验测得IR谱分布区间相对吻合, 三种物质的IR振动波峰均分布在200~2 000和2 000~4 000 cm-1区间, 并且处于200~2 000 cm-1之间的波峰数目多, 而处于2 000~4 000 cm-1区间波峰数目少, 即整个振动谱分布于中红外(400~4 000 cm-1, 2.5~25 μ m)和远红外(10~400 cm-1, 25~1 000 μ m)区域, 表明处于中红外区域的光谱属于T2, T3, T4分子的基频振动谱(例如, T3分子在3 145 cm-1处振动峰值是由于苯基上的C— H键之间的非对称伸缩振动引起的), 处于远红外区域的光谱属于分子的转动光谱和某些集团的振动光谱(例如, T3分子在209 cm-1处振动峰值是由于苯基与氧原子之间的剪式振动以及骨架转动引起的)。 T2', T3', T4'的IR谱与T2, T3, T4的IR谱相比, T2', T3', T4'的振动谱出现展宽以及振动强度与T2, T3, T4的振动谱有所差异, 这是由于使用KBr压片法测量时引入的KBr吸湿性较强, 特别是对含有O— N、 N— H、 C=C、C=N键伸缩振动会造成干扰, 其次样品在压片过程中会发生物理变化(如多晶转换现象)或化学变化(部分分解), 使图谱面貌出现差异。 T4″的IR谱与T4的IR谱相比, 同样也出现展宽, 这是由于使用石蜡糊法测量时引入的石蜡本身属于有机物, 并且石蜡自身具有一定的IR谱, 因此对图谱结果也会造成一定影响。 理论计算环境相对单一, 可以排除上述因素, 相对合理的对IR谱位置分布以及振动强度作出预判, 从上述分析得出所选计算方法具有可行性和准确性。
 | 图2 T2, T3, T4的红外光谱Fig.2 IR spectra of T2, T3, T4 |
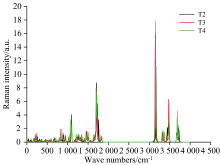
图3给出了T2, T3, T4分子的拉曼(Raman)振动谱, 黑色波谱、 红色波谱、 绿色波谱分别对应T2, T3, T4分子。 由图3可看出, 三种物质的Raman振动波峰均分布在0~2 000和2 000~4 000 cm-1区间, 并且处于0~2 000 cm-1之间的波峰数目多, 而处于2 000~4 000 cm-1区间波峰数目少, 即整个振动谱分布于中红外(400~4 000 cm-1, 2.5~25 μ m)和远红外(10~400 cm-1, 25~1 000 μ m)区域, 表明处于中红外区域的光谱属于T2, T3, T4分子的基频振动谱(例如, T3分子在3 145 cm-1处振动峰值是由于苯基上的C— H键之间的非对称伸缩振动引起, 1 093 cm-1处振动峰值是由于分子上所有C— C键之间的剪式振动引起的, 1 705 cm-1处振动峰值是由于苯基上的C— C键之间的拉伸振动以及C— H键之间的非平面摇摆振动引起的, 3 478 cm-1处振动峰值是由于O— H键之间的非平面摇摆振动引起), 处于远红外区域的光谱属于分子的转动光谱和某些集团的振动光谱(例如, T4分子在248 cm-1处振动峰值是由于苯基与苯基之间的非对称剪式转动引起的)。
 | 图3 T2, T3, T4的拉曼光谱Fig.3 Raman spectra of T2, T3, T4 |
结合图2和图3得出, T2、 T3、 T4团簇的IR和Raman振动谱分布区间相同(均分布在0~2 000和2 000~4 000 cm-1区间), 包括振动强度也相似, 这为实验方面测定T2、 T3、 T4物质的Raman光谱提供了参照。 由上述分析可得出, T2、 T3、 T4团簇的IR和Raman振动谱分布于中红外和远红外区间。 各分子内部振动模式对IR和Raman振动峰值的分布具有重要的影响, C15NO4HnIm (n=11, 12, 13, m=4, 3, 2; n+m=15)分子的基频振动促使振动谱分布于中红外区; C15NO4HnIm (n=11, 12, 13, m=4, 3, 2; n+m=15)分子的转动光谱和分子内特定集团的振动促使振动谱分布于远红外区域。
用高斯曲线拟合的T2, T3, T4在水溶剂下的吸收谱如图4所示, 即黑色波谱、 红色波谱、 绿色波谱分别对应T2, T3, T4分子。 由图可以看出, T2分子具有两个吸收峰, 低能量的吸收峰(> 325 nm)是由于苯环上原子之间的π — π * 键过渡引起的, 高能量的吸收峰(200~300 nm)主要是由于氧原子以及碘原子与苯环之间的π — π * 键过渡引起的。 同样, T3分子具有两个吸收峰, 低能量的吸收峰(> 350 nm)是由于苯环上原子之间的π — π * 键过渡引起的, 高能量的吸收峰(200~325 nm)是由于氧原子以及碘原子与苯环之间的π — π * 键过渡引起的。 T4分子具有两个吸收峰, 低能量的吸收峰(> 375 nm)主要是由于苯环上原子之间的π — π * 键过渡引起的, 高能量的吸收峰(200~375 nm)是由于氧原子以及碘原子与苯环之间的π — π * 键过渡引起的。 随着分子含碘数量的增多, 吸收谱的振子强度逐渐减小, 说明碘原子数量的增多会影响分子内部电子跃迁, 随着I取代苯环上H原子的数量增多, 会抑制分子内部电子跃迁, 也会抑制氧原子以及碘原子与苯环之间的π — π * 键过渡。
 | 图4 T2, T3, T4在H2O溶剂下的吸收谱Fig.4 Absorption spectra of T2, T3, T4 in H2O solution |
表4列出了图4中相应峰值的能量跃迁出处。 例如, 图4中T2的高能吸收峰吸收能量主要是由于HOMO-2→ LUMO+4(27.1%)、 HOMO-1→ LUMO+2(39.8%)和HOMO-1→ LUMO+4(20.3%)的轨道电子跃迁产生的, 低能吸收峰396.72 nm处的能量主要是由于HOMO-1→ LUMO(100%)的轨道电子跃迁产生的。 T3的高能吸收峰吸收能量主要是由于HOMO-1→ LUMO+3(67%)和HOMO-2→ LUMO+7(14%)的轨道电子跃迁产生的, 低能吸收峰402.42 nm处的能量主要是由于HOMO-1→ LUMO(100%)的轨道电子跃迁产生的。 T4的高能吸收峰吸收能量主要是由于HOMO-4→ LUMO(86%)和HOMO-4→ LUMO+1(9.5%)的轨道电子跃迁产生的, 低能吸收峰445.85 nm处的能量主要是由于HOMO→ LUMO+1(80%)的轨道电子跃迁产生的。
| 表4 用TDDFT计算得到T2, T3, T4在H2O溶剂中的吸收特性 Table 4 Absorption properties of T2, T3, T4 in H2O solution obtained with the TDDFT calculations |
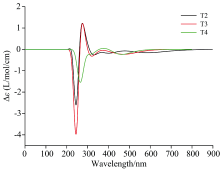
用高斯曲线拟合的T2, T3, T4在水溶剂下的电子圆二色性(ECD)发射谱如图5所示, 即黑色波谱、 红色波谱、 绿色波谱分别对应T2, T3, T4分子。 由图5可以看出, 三种团簇波谱的波峰值既有正值又有负值, 并且都在紫外可见光波长范围内, 纵坐标为正值, 即为右旋, 纵坐标为负值, 即为左旋, 对于这种峰值有正负的现象是由于手性中心的不同所引起的。 例如, T2有两个显著波峰位于244和274 nm, 244 nm处纵坐标为负值(-2.60左旋), 274 nm处纵坐标为正值(1.22右旋); 同样, T3也有两个显著波峰分别位于244.8 和275.2 nm, 244.8 nm处纵坐标为负值(-3.97左旋), 275.2 nm处纵坐标为正值(1.21右旋); 这两种团簇的其余波峰则围绕零点处横坐标小幅振动. 相对应的T4发射谱具有一个显著发射峰, 位于265.6 nm处的峰值均为负值(-1.54左旋), 该团簇的其余波峰则围绕零点处横坐标小幅振动。
 | 图5 T2, T3, T4在H2O溶剂下的ECD谱Fig.5 ECD spectra of T2, T3, T4 in H2O solution |
基于密度泛函理论(DFT)在B3LYP/Lanl2mb基组水平, 研究了优化后的T2, T3, T4团簇的基态性能(点群、 结合能、 NIMAG、 能隙、 电离势、 电子亲和能、 提取势和重组能等)。 结果表明, T2, T3, T4团簇基态结构对称性为C1; T2分子既不具有p型输运性质也不具有n型输运性质, T3和T4分子均具有p型输运性质. 这三种团簇的IR和Raman振动谱分布于中红外和远红外区间, 各分子内部振动模式对IR和Raman振动峰值的分布具有重要影响, C15NO4HnIm (n=11, 12, 13, m=4, 3, 2; n+m=15)分子的基频振动促使振动谱分布于中红外区; C15NO4HnIm (n=11, 12, 13, m=4, 3, 2; n+m=15)分子的转动光谱和分子内特定集团的振动促使振动谱分布于远红外区域。 运用含时密度泛函理论(TDDFT)同样在B3LYP/Lanl2mb基组水平上研究发现, 三种团簇在水溶剂下的吸收谱都有两个显著峰值, 其中低能吸收峰主要是由于苯环上原子之间的π — π * 键过渡引起的, 而高能吸收峰主要是由于氧原子以及碘原子与苯环之间的π — π * 键过渡引起的; 三种团簇的手性ECD发射谱在紫外可见光区域内既有左旋又有右旋, 是由于手性中心的不同所引起的。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|