

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
氟取代对香豆素分子超快动力学过程的影响
[葛晶1, 2  , 李智彪
, 李智彪1, 2 , 薛丙乾1, 2 , 白西林1, 2, * ]
, 李智彪]
|
|


作者简介: 葛 晶, 女, 1987年生, 山西师范大学物理与信息工程学院讲师 e-mail: 703366@sxnu.edu.cn
设计具有高亮度、 高光稳定性和环境友好型的荧光染料, 是当前备受关注的前沿领域。 研究发现, 荧光染料的发光效率受到扭转分子内电荷转移(TICT)的影响。 因此, 有效地抑制TICT过程对于荧光标记物和探针的研究与开发具有至关重要的意义。 香豆素及其衍生物, 尤其是具有高荧光产率和长荧光寿命的7-氨基香豆素染料, 一直以来都广泛应用于不同体系中, 作为荧光探针的重要组成部分。 然而, 以往的研究主要集中在探究不同溶剂对香豆素染料分子激发态动力学过程的影响, 而忽略了染料分子结构对TICT过程的重要作用。 采用了飞秒时间分辨瞬态吸收(TA)光谱和密度泛函理论(DFT)计算相结合的方法, 深入研究香豆素染料C460和C481在不同溶剂中的激发态动力学。 研究结果揭示, 在具有高强供氢能力的高极性甲醇溶剂中, C481才显著发生TICT过程。 此外, 量子化学计算结果表明, 氟取代效应会降低分子内扭转势垒, 导致非辐射失活效应加速, 从而引发更为明显的荧光猝灭现象。 这一研究不仅有助于优化荧光染料性能时选择合适的溶剂, 还可为荧光染料分子结构的设计提供重要的指导, 具有深远的研究价值。
The development of highly efficient, photostable, and eco-friendly fluorescent dyes is currently a prominent focus in scientific research, attracting significant attention. Recent studies have emphasized Twisted Intramolecular Charge Transfer (TICT) 's crucial role in determining fluorescent dyes' luminescence efficiency. Consequently, effectively suppressing the TICT process is imperative for progressing fluorescent markers and probes. Among the various fluorescent dyes, coumarin and its derivatives, particularly 7-amino coumarin dyes, have gained significant recognition as extensively utilized constituents in diverse systems owing to their robust fluorescence and prolonged fluorescence lifetimes. Nevertheless, previous examinations have predominantly concentrated on the effect of different solvents on the excited-state behavior of coumarin dye molecules, overlooking the significant influence of dye molecule structure on the TICT process. This study utilized a combination of femtosecond time-resolved transient absorption (TA) spectroscopy and density functional theory (DFT) calculations to gain deeper insights into the excited-state dynamics of coumarin dyes C460 and C481 in various solvents. The results revealed that C481 predominantly undergoes the TICT process in highly polar methanol solvents with strong hydrogen-bonding capabilities. Furthermore, quantum chemical calculations indicated that introducing fluorine substitutions reduces the molecules' internal torsional barriers, leading to enhanced non-radiative deactivation processes and, consequently, more pronounced fluorescence quenching phenomena. This investigation provides insights into the selection of suitable solvents for optimizing the performance of fluorescent dyes and offers valuable guidance for their design.
设计高亮度、 高光稳定性以及对环境敏感的荧光染料, 是当前备受关注的热门研究领域。 其中, 扭转分子内电荷转移(twisted intramolecular charge transfer, TICT)一直是研究者们探索的光物理现象。 TICT最初由Grabowski等提出, 用以解释p-n, n-二甲氨基苯腈(DMABN)分子所表现出的双荧光发射峰现象[1]。 当具有TICT特征的分子受到光激发时, 分子内电子给体(D)或受体片段(A)会环绕单键或双键进行扭转[2]。 这个过程导致了分子的荧光被明显猝灭, 并会显著降低它们的光稳定性。 因此, 如何有效地抑制TICT过程对于研究和开发荧光标记物以及荧光探针具有极其重要的研究意义[3, 4, 5]。
香豆素及其衍生物属于一类重要的有机荧光染料, 研究表明它们的激发态动力学过程容易受到溶剂的极性、 粘度以及分子间氢键[6, 7, 8, 9, 10]的影响。 在众多香豆素衍生物中, 7-氨基香豆素染料表现出高荧光量子产率和长荧光寿命。 然而, 在光激发下, 大多数7-氨基香豆素染料的分子内偶极矩会发生明显的变化, 导致吸收和发射光谱中出现显著的斯托克斯位移现象。 这一现象可归因于分子内电荷转移过程, 即电子从电子给体7-氨基基团转移到电子受体基团。 香豆素460(C460)是一种典型的7-氨基香豆素染料, 广泛作为荧光探针掺杂到不同的体系中, 并且已经用许多实验方法进行了详细研究。 Atanu Barik等通过测量C460在不同极性溶剂中的荧光量子产率, 提出了C460在高极性溶剂(溶剂极性参数Δ f> 0.3)中存在一个独特的动力学过程, 将其归属于TICT过程[11]。 随后, 李尤等利用密度泛函理论(density functional theory, DFT) 和含时密度泛函理论(TDDFT)研究了分子间氢键对C460激发态动力学过程的影响, 发现分子间氢键的相互作用促进了TICT态的形成, 从而导致C460在甲醇溶剂中的荧光强度明显低于乙腈溶剂[12]。 通常染料分子结构的改变会显著影响其激发态动力学过程, 比如荧光猝灭。 其中, 为了探究氟元素取代对ICT过程的影响, Mhejabeen Sayed对香豆素-102 (C102) 与香豆素-153 (C153) 进行研究[13], 说明了C153染料的全氟甲基(— CF3)的存在会使其在低极性溶剂中的非辐射退激发作用增强, 从而导致更强的荧光猝灭现象。 并且量化计算结果表明这一非辐射过程为隙间穿越(intersystem crossing, ISC) 过程。 然而, 早期研究未考虑分子结构的改变是否会对TICT过程产生影响。
选取了两种结构相似的香豆素分子作为研究对象, 即C460和C481, 深入研究了它们在不同溶剂中的激发态动力学。 这两种分子均具有相同的氢键受体, 可以与供体溶剂甲醇(MeOH)、 正丁醇(BA)和乙腈(ACN)形成分子间氢键, 唯一区别在于它们苯环上的基团, 一个是甲基(— CH3), 另一个是氟甲基(— CF3)。 首先, 通过飞秒瞬态吸收(fs-TA)光谱观测了这两种分子激发态下的超快动力学过程。 随后, 借助DFT和TDDFT对C460和C481在三种不同溶剂中形成的氢键复合物进行了基态(S0)和第一电子激发态(S1)的几何构型的优化, 同时进行了静电势分析、 电子光谱计算、 前线分子轨道分析和激发态势能面扫描。 通过以上实验观测和理论计算, 将揭示氟取代如何引起C460和C481分子在激发态动力学过程中的差异。 这项研究将为设计和开发高灵敏度荧光探针提供重要的指导意义。
研究用的药品包括C481、 MeOH、 ACN和BA, 均从Aladdin试剂公司购买。 C460购自Exciton公司。 所有实验药品皆未经进一步纯化处理。 在实验中, 以C460和C481作为溶质, 另外三种试剂作为溶剂, 制备了6组浓度为100 μ mol· L-1的实验样品, 并将其分装到1 mm光程的石英玻璃容器中, 以进行稳态/瞬态光谱实验测量。
首先, 使用FluoroMax+荧光光谱仪(Horiba)获取了实验样品的稳态吸收光谱和发射光谱。 接着, 使用钛蓝宝石激光器Astrella(Coherent)和飞秒泵浦-探测系统Transpec-FS(中智科仪)获得了飞秒瞬态吸收光谱。 具体来说, 激光放大器产生的飞秒激光(800 nm, 1 kHz, 35 fs)按照7∶ 3的比例分成两束, 分别作为泵浦光和探测光。 其中较强光束经过光参量放大器TOPAS-Prime(Coherent)产生泵浦光(375 nm), 用于激发实验样品。 较弱光束通过光学延迟线(0~4 ns)聚焦在CaF2窗口上, 产生350~750 nm的连续白光作为探测光。 通过调节光斑空间位置以确保两束光在实验样品上重叠, 并使用斩波器(1 kHz)对泵浦光进行斩波, 从而获得样品在有和无泵浦光时所引起的吸收变化(Δ A/MOD)。 所有实验均在常温常压的环境中进行。
使用Gaussian09软件对C460和C481单体以及它们的氢键复合物进行了高精度的DFT和TDDFT计算[14]。 首先, 采用B3LYP/TZVP方法和基组用于基态和激发态的几何构型优化、 频率分析、 静电势分析、 垂直激发能、 荧光发射能、 振子强度和前线分子轨道分析。 同时, 为了更准确地考虑溶剂效应, 采用可极化连续介质模型(IEFPCM)进行计算[15]。 其次, 为验证TICT态的产生, 构建了S1态的势能面(PESs)。 为了更准确的预测TICT态的能量和性质, 在PESs计算中采用了CAM-B3LYP/TZVP方法和基组, 并采用cLR-SMD溶剂模型[16]。
为了考察溶剂极性和氢键形成对TICT过程的影响, 这里选择了三种不同的溶剂, 并结合极性参数Δ f(精确描述极性)和K-T理论(准确表征供氢性能)。 这些溶剂的详细参数如表1所示: Δ f值大于0.28的溶剂被视为高极性溶剂; 而较高的α 值表示溶剂具有更强的供氢性能, 从而与溶质分子形成更强的氢键。 Δ f和Kamlet-Taft(K-T)的数学表达式如式(1)
式(1)中: ε 为溶剂静态介电常数; n为示折射率[17]。
式(2)中: α 为供氢能力; β 为受氢能力; π * 为溶剂的极性或极化率[18]。
| 表1 MeOH、 ACN和BA的溶剂参数Δ f和K-T参数(α 、 β 、 π * ) Table 1 Solvent parameters Δ f and K-T parameters (α , β , π * ) for MeOH, ACN, and BA |
由于K-T理论中的π * 是多元线性回归方程的众多参数之一, 我们认为对于溶剂极化率的描述没有极性参数中的溶剂极化率方程精确。 因此我们选择准确性更高溶剂极性参数Δ f去分析溶剂的极性。
溶剂极化率方程
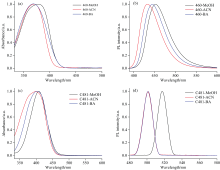
C460和C481在不同溶剂中的稳态吸收和荧光发射光谱, 如图1所示。 相应的吸收峰和发射峰以及斯托克斯位移值, 如表2所示。 通过比较发现, C460和C481在不同溶剂中的斯托克斯位移的大小排序与氢键复合物的分子间氢键强度排列一致, 也与溶剂分子供氢能力α 值的大小排序相一致, 即从大到小的顺序是MeOH> BA> ACN。
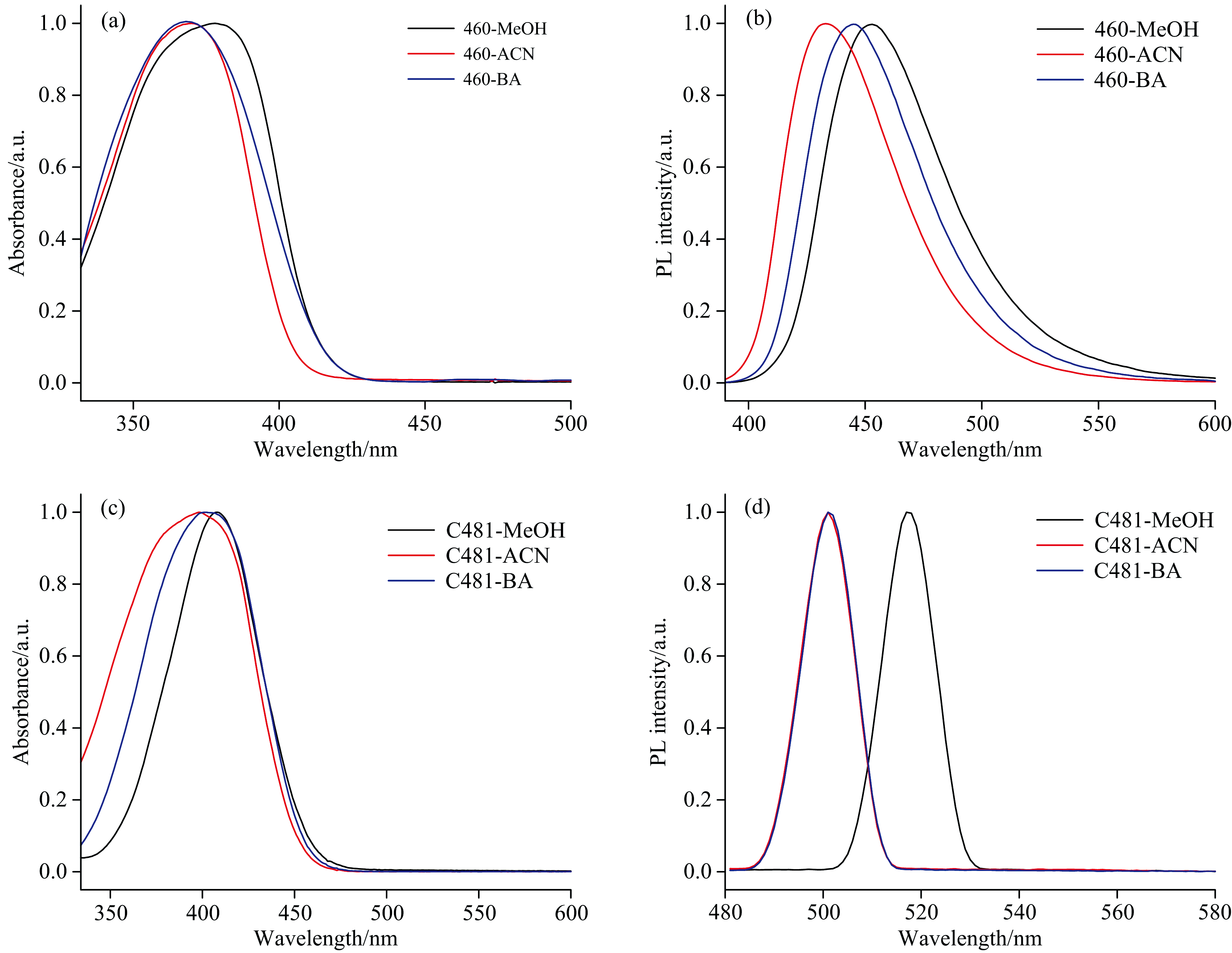 | 图1 C460/C481在MeOH、 ACN和BA溶剂中的稳态吸收(a, c)和荧光发射(b, d)光谱图Fig.1 Steady-state absorbance (a, c) and fluorescence emission (b, d) spectra of C460/C481 in MeOH, ACN, and BA |
| 表2 C460和C481在MeOH、 ACN和BA中的吸收和发射峰以及斯托克斯位移(nm) Table 2 Absorbance and emission peaks and corresponding stokes shift values (nm) of C460 and C481 in MeOH, ACN, and BA |
在375 nm波长的光激发下, C460和C481在MeOH、 ACN和BA溶剂中的fs-TA光谱如图2所示。 观察发现, 在0~200 ps的时间延迟范围内, C460在这三种溶剂中, 400~425 nm波长区域呈现的瞬态吸收(transient absorption, TA)正信号可归属为激发态吸收(ESA)。 在相同延迟时间段内, 最强的负信号对应波长在~450 nm, 可将其归属为受激发射(SE)。 而在1 ps以内, 出现在~425 nm的负信号可归属为基态漂白信号(GSB)。 相比之下, C481复合物的TA正信号范围更宽, 为400~450 nm, SE的峰值出现在~500 nm。 最有趣的是, C481-MeOH复合物的最大发射峰表现出480~525 nm的显著红移特征, 伴随着SE带范围的增加, 表明存在一个与ICT过程不同的动力学过程。 通过后续的理论计算, 这一动力学过程被归属于TICT态的形成。
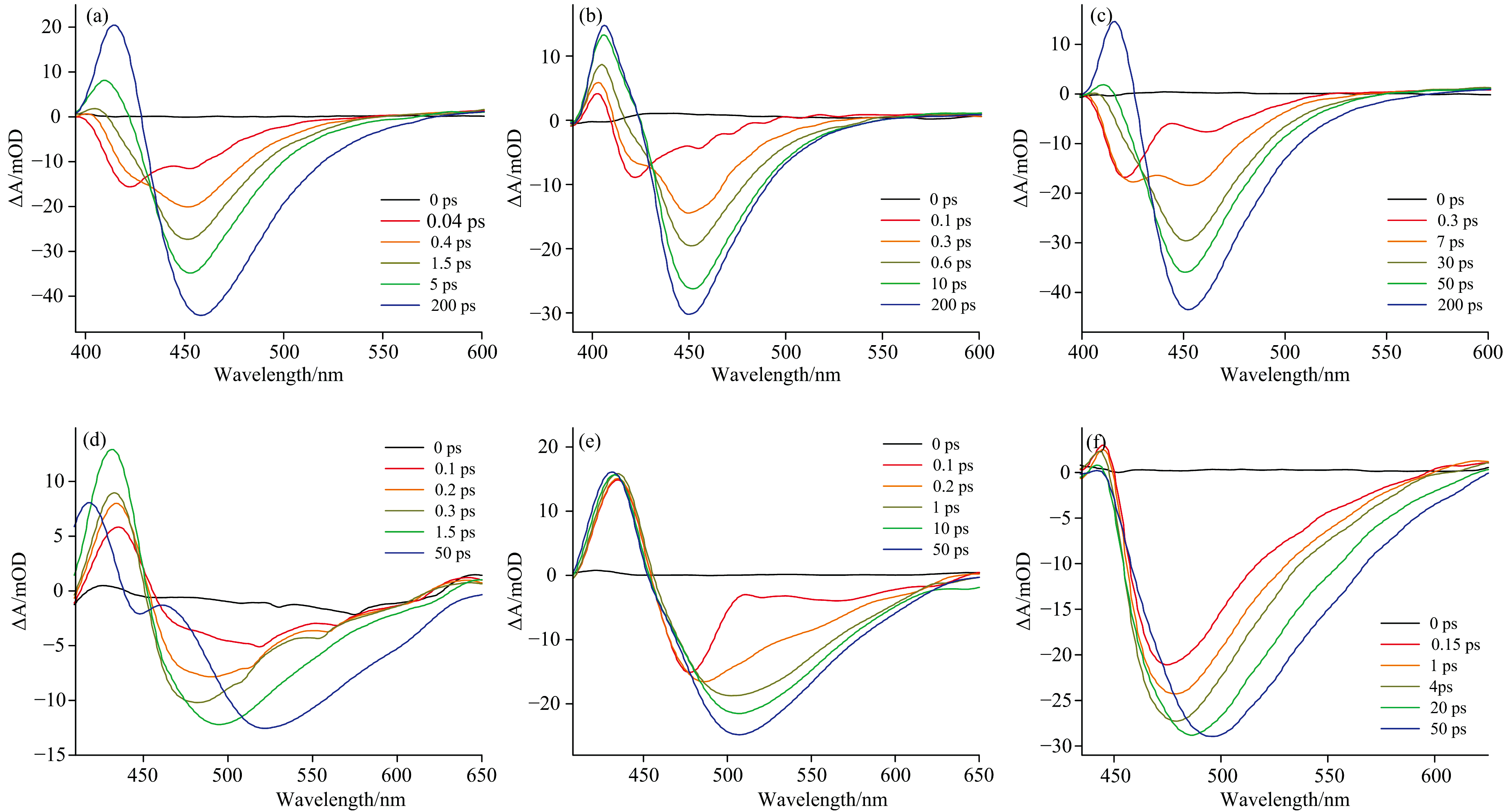 | 图2 C460/C481在MeOH(a, d)、 ACN(b, e)和BA(c, f)中不同时间延迟下的fs-TA光谱Fig.2 Transient absorption spectra of C460/C481 in MeOH (a, d), ACN (b, e), and BA (c, f) solvents at different delay times |
为了进一步分析氢键复合物激发态动力学过程, 对最大发射峰波长处的动力学曲线进行了全局拟合分析, 如图3和表3所示。 除C481-MeOH需要四指数函数拟合外, 其余都可以很好地通过三指数函数拟合。 在这些拟合参数中, 参数τ 1对应激发态的ICT动力学过程, 表3中可以看到C460在MeOH溶剂中的τ 1为2.23 ps, 而C481在MeOH溶剂中的τ 1大幅缩短为0.28 ps, 另外在BA溶剂中也从6.5 ps(C460)减小到了1.32 ps(C481), 由此可以说明氟取代加快了ICT过程的速率; ~2 ps左右, SE峰值开始上升, 在~40 ps时达到最大值, 然后开始衰减, 时间尺度~ns。 2 ps的时间点正好对应光谱图上SE带的红移, 这是由于光激发后C481分子中发色团的超快溶剂化。 根据DFT计算结果, 可以将这一过程归属于TICT态的形成。 在SE峰值达到最大值后, 还有两个缓慢的衰减过程。 根据先前的研究, 可以将这两个时间常数分别归属为溶剂化过程和荧光发射过程, 分别在数十皮秒(τ 3)和纳秒级(τ 4)时间尺度内发生[19]。
 | 图3 C460/C481在MeOH(a, d)、 BA(b, e)和ACN(c, f)中的TA动力学曲线Fig.3 Kinetic parameters of C460 and C481 in MeOH, ACN, and BA obtained by global fitting |
| 表3 全局拟合得到C460和C481在MeOH、 ACN和BA中的动力学参数 Table 3 Kinetic parameters of C460 and C481 in MeOH, ACN, and BA obtained by global fitting |
综上所述, 通过比较相同溶剂中不同溶质分子的激发态动力学过程, 可以得出结论: 氟取代显著提高了分子内电荷转移的效率[13]。 另一方面, 下文的理论计算表明氟取代降低了TICT的扭转势垒, 因此在甲醇溶剂中, C481相较于C460更容易发生TICT过程, 导致更强的荧光猝灭现象。 而通过比较不同溶剂中相同溶质分子的激发态动力学过程可以得出: 分子间氢键对ICT过程的影响明显高于极性因素, 但仅在同时满足高极性和溶质与溶剂分子间形成较强分子间氢键的情况下, C481才能形成稳定的TICT态。
2.3.1 静电势分析
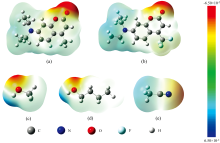
为了了解溶质与溶剂分子之间是否会形成氢键以及其形成氢键位置, 对C481和C460以及溶剂分子进行静电势分析, 如图4所示。 此图能够将一个分子官能团的电荷分布和电负性形象的展现出来, 红色代表分子结构中高电负性区域, 蓝色代表低电负性区域, 高-低电负性区域相互吸引就会形成分子间氢键。 从图中可以看到: C481和C460分子结构中的C=O位置呈现亮红色, 表明此处电负性最强; 而MeOH和BA中O— H基团和ACN中的C— H基团都由蓝色覆盖, 说明三者所处区域电负性最弱。 综上所述, 溶剂中亲电基团的H原子可以作为氢键供体, 与溶质分子中的氢键受体C=O键形成分子间氢键。 根据分析结果, 可以得出结论: C481和C460在MeOH、 ACN和BA溶剂中均会形成分子间氢键, 再结合K-T理论中α 值推测, 强弱排序为MeOH> BA> ACN。 通过对比C460和C481分子的静电势图, 可以明显发现两者在右下角的电势分布有所区别: C481中全氟甲基的颜色要比C460中甲基部分更绿, 对比静电势色条来看, 说明要比甲基的电负性更强。 这也就导致C481分子结构中C=O位置的电负性相对来说偏弱, 使得C481与三种溶剂分子形成的氢键强度弱于C460。 此结论将在几何构型优化中进一步论证。
 | 图4 C460(a)、 C481(b)、 MeOH(c)、 BA(d)和ACN(e)分子的ESP图Fig.4 ESP maps of C460 (a), C481 (b), MeOH (c), BA (d), and ACN (e) molecules |
2.3.2 几何构型优化
使用B3LYP/TZVP计算水平对C460和C481以及它们与三种不同溶剂分子形成的氢键复合物的S0和S1态几何构型进行了优化。 C481-MeOH复合物在基态和第一电子激发态下的几何构型优化结果, 其中关键的键长参数如表4所示。
| 表4 C460和C481氢键复合物的重要键长信息(Å ) Table 4 Key bond length (Å ) information of the hydrogen-bonding complexes of C460 and C481 |
以C481为例, 在S0态下, C481与MeOH、 BA以及ACN形成的氢键的键长分别为1.87、 1.88和2.35 Å , 这表明氢键的长度与溶剂的α 值呈线性正相关。 值得注意的是, C481-MeOH氢键长度1.879 Å 大于C460-MeOH氢键长度1.847 Å , 说明C481分子与MeOH溶剂分子形成的氢键强度小于C460分子, 在其他溶剂中也是如此, 这与之前进行的静电势分析结果一致。 此外, 在S1态下, 相应的氢键长度都有所缩短, 表明分子间氢键在激发态下得到了增强。 C460的相关参数显示出的规律与以上描述一致, 因此不再做具体分析。
2.3.3 垂直激发能与电子光谱
通过计算获得了C460和C481分子单体以及在不同溶剂中的垂直激发能以及相应的振子强度, 如表5所示。 研究结果表明, 所有氢键复合物的S1态均对应于表中的最大振子强度, 说明S1态的吸收在吸收光谱中占主导地位。 与单体相比, 复合物的垂直激发能出现明显的红移现象, 这证明了分子间氢键的形成有效地降低了基态与激发态之间的能级差。 此外, 稳态光谱测量结果也印证了这一观点。
| 表5 C460和C481单体以及氢键复合物的垂直激发能(nm)及相应的振子强度 Table 5 Vertical excitation energies (nm) and corresponding oscillator strengths of C460 and C481 monomers, as well as their hydrogen-bonded complexes |
2.3.4 前线分子轨道分析
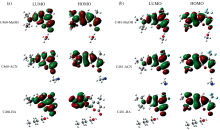
为了可视化激发态ICT过程, 对氢键复合物的前线分子轨道进行了分析, 如图5所示。 研究结果表明, HOMO到LUMO轨道的跃迁对应于S1态, 具有最强的振子强度。 从HOMO跃迁到LUMO轨道, 氢键复合物的电子密度经历了重新分布: 原本分布在左侧乙基部分的电荷被转移到了右侧的羰基上; 原本位于右侧的甲基和三氟甲基在跃迁后也有少量电荷分布。 简而言之, 当C460和C481氢键复合物被激发到S1态时, 分子内电荷经历了重新排布, 即电荷发生了转移, 也就意味着氢键复合物在S1态具有显著的ICT特性。
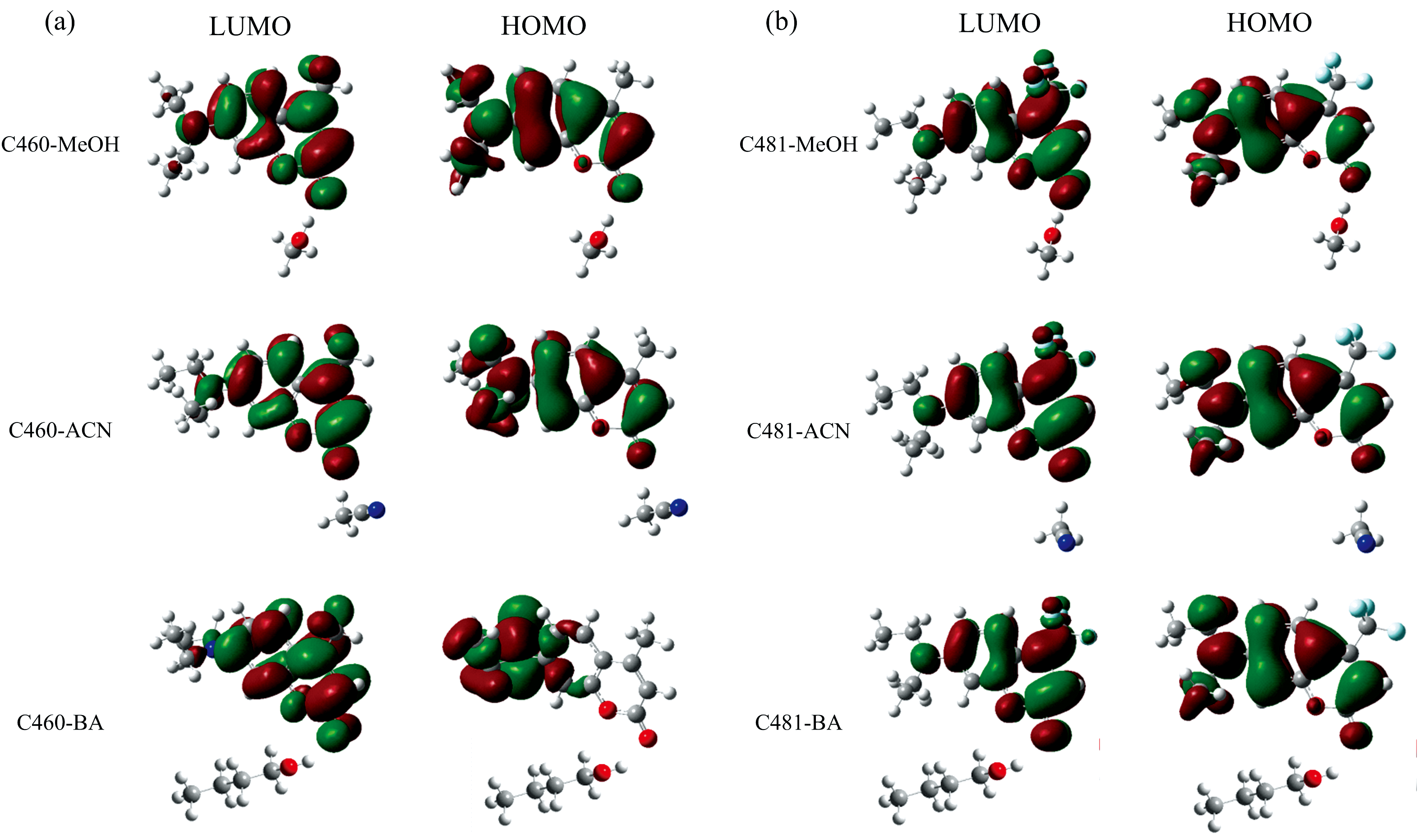 | 图5 C460和C481单体以及氢键复合物的垂直激发能(nm)及相应的振子强度2.3.5 激发态势能面扫描Fig.5 Vertical excitation energies (nm) and corresponding oscillator strengths of C460 and C481 monomers, as well as their hydrogen-bonded complexes |
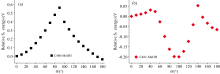
为了详细分析氟取代对C460和C481在甲醇溶液中激发态动力学的影响, 采用CAM-B3LYP/TZVP基组和泛函并利用cLR-SMD溶剂模型计算了两者的势能面, 如图6所示。 其中, 水平坐标表示7-氨基基团与苯环平面的二面角, 垂直坐标表示氢键复合物相对于S1态的能量。 从图中明显可以观察到, C460分子在甲醇溶剂中, 随着7-氨基基团的扭转, 相对于S1态的能量逐渐增加, 达到了0.56 eV的扭转势垒。 相比之下, C481分子只需跨越约0.031 eV的势垒, 然后在扭转角接近90° 的位置达到了总能量的最小值。 通过这两种复合物势能面的对比发现, C460分子中的氢原子被氟原子取代后, 降低了TICT过程发生所需的扭转势垒, 因此更容易发生荧光猝灭现象, 这也是两者激发态动力学过程产生差异的主要原因。
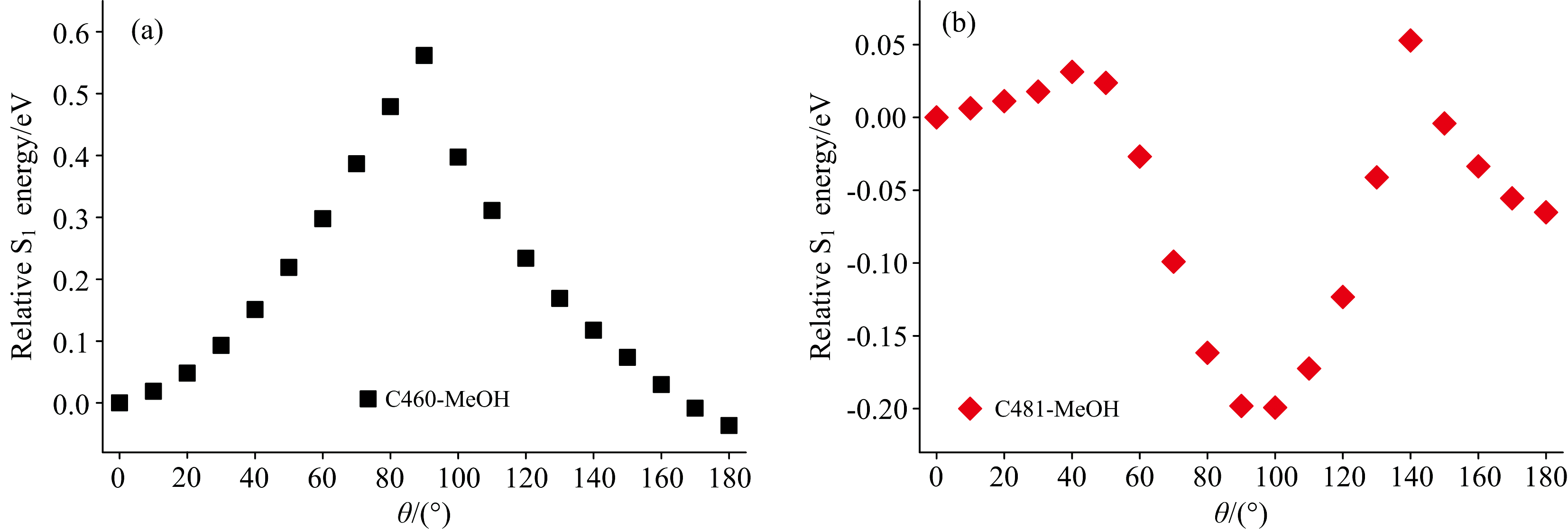 | 图6 C460和C481在甲醇溶剂中形成的氢键复合物S1态势能面Fig.6 The potential energy surface of hydrogen bond complex formed by C460 (a) and C481 (b) in MeOH solvent at S1 state |
采用稳态/瞬态吸收光谱和密度泛函理论计算相结合的方法, 深入研究了C460和C481染料分子在不同溶剂中的激发态动力学过程。 具体而言, C481分子只有在高极性溶剂(具有较高Δ f值)中, 并且存在强分子间氢键的条件下才会发生TICT过程。 量化计算结果显示, 分子间氢键的强度与K-T理论中的α 值呈正相关关系。 并且激发态氢键的显著增强促使了TICT态的形成。 另一方面, C460分子中的甲基氢原子被氟原子取代后, 显著改变了分子内电荷分布, 从而降低了形成TICT态的扭转势垒。 这意味着氟取代效应引入了新的非辐射失活通道, 导致非辐射失活效率更高, 荧光猝灭现象也更加显著。 这些研究结果将有助于新型荧光染料的分子结构设计。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|