
{kind=link}
{kind=link}
基于密度泛函理论的沙林毒剂近红外光谱研究
[孙梽珅1  , 刘永刚
, 刘永刚2, 3 , 张旭1 , 郭腾霄1, * , 曹树亚1, * ]
, 刘永刚, 曹树亚|
|


作者简介: 孙梽珅, 1992年生, 国民核生化灾害防护国家重点实验室硕士研究生 e-mail: sunzhishen@foxmail.com
近红外光谱涵盖了有机分子中C—H, N—H和O—H等含氢基团的倍频和合频产生的光谱, 提供了分子的结构、 组成、 状态等信息, 是研究有机物含氢基团振动的重要方法, 常用于食品、 农作物等的定性定量分析。 生化领域内所研究对象也都含有氢基团, 这些含氢基团吸收频率特征性强, 受分子内外环境影响小, 近红外光谱特性更稳定, 故可用于化学战剂和危险化学品检测。 沙林是一种神经性化学毒剂, 研究其结构、 化学特性及光谱性质时, 为保证安全, 实验中常用模拟剂样品代替测试, 但目前尚无公允的沙林毒剂近红外模拟剂。 采用密度泛函理论(DFT), 基于Gaussian程序包, 利用B3LYP/def2-SVP对沙林分子进行基态结构优化, 计算了沙林分子的精细结构和分子基频振动模式, 引入广义二阶微扰理论(GVPT2)建立了模拟生化毒剂近红外光谱的理论模型, 得到近红外振动峰与主要振动模式, 由倍频(Overtones)和合频(Combination Bands)振动绘制得到近红外光谱。 对沙林在近红外区域内的含氢基团进行解析, 对其特征峰进行指认, 得到沙林分子在1 150、 1 362和1 500 nm处的三个特征峰及其振动模式, 其中1 150 nm峰是由多个倍频和合频的组合振动贡献产生; 1 362 nm是一个较宽的吸收振动峰, 主要由分子中与C原子相连的原子合频和其他的非C, H原子产生的倍频或合频引起的; 1 500 nm位置的近红外振动峰主要由C8相关的振动模式贡献产生。 通过密度泛函理论建立沙林的近红外光谱理论模型, 通过实验验证了其理论模型的可行性, 为寻找其近红外光谱模拟剂提供理论支撑。
Near-infrared spectroscopy is mainly the overtones and combination bands absorption spectra of organic molecules, which are generated by the overtones and combination bands of hydrogen-containing groups such as C—H, N—H, O—H, etc., which can obtain molecular structure, composition, state and other information. This technology is an important method for studying the vibration information of hydrogen-containing groups in organic matter and is often used for qualitative and quantitative analysis of biological substances such as food and crops. The research objects in the biochemical field also have hydrogen-containing groups. These hydrogen-containing groups have strong absorption frequency characteristics, are less affected by the internal and external environment of the molecule, and have more stable spectral characteristics in the near-infrared spectrum. This technology can be used to detect chemical warfare agents and hazardous chemicals. Sarin is a neurotoxic chemical agent. When studying its structure, chemical properties and spectral properties, in order to ensure safety, simulants are often used in the experiment to substitute for testing, but there is no fair near-infrared simulant for sarin. This paper uses density functional theory (DFT), based on the Gaussian program package, and uses B3LYP/def2-SVP to optimize the ground state structure of the sarin molecule, and calculates the fine structure of the sarin molecule and the fundamental frequency vibration mode of the molecule. The generalized second-order perturbation theory (GVPT2) is introduced to establish a theoretical model for simulating the near-infrared spectrum of biochemical poisons, obtaining the near-infrared vibration peaks and main vibration modes, and the near-infrared spectrum drawn from the vibrations of overtones and combination bands. Analyze the hydrogen-containing groups of sarin in the near-infrared region, use this method to identify its characteristic peaks, obtain three characteristic peaks of sarin molecules at 1 150, 1 362 and 1 500 nm and analyze their vibration modes. Among them, the position at 1 150 nm is produced by the contribution of multiple overtones and combination bands vibration. 1 362 nm is a wide absorption vibration region, mainly caused by the combination bands of atoms connected to C atoms in the molecule and other non-C, H atoms. The near-infrared vibration peak at 1 500 nm is mainly caused by the C8 Related vibration mode contribution. In this paper, the theoretical model of Sarin's near-infrared spectroscopy is established through density functional theory, and the feasibility of the theoretical model is verified through experiments, which provides theoretical support for finding its near-infrared spectroscopy simulation agent.
光谱分析是根据物质的光谱来鉴别物质种类, 确定其化学组成和相对含量的一种常用方法。 近红外光(near infrared, NIR)是介于紫外-可见光(ultraviolet-visible, UV-Vis)和中红外光(mid-infrared, MIR)之间的电磁辐射波, 波长范围为700~2 500 nm(1 4286~400 cm-1)[1]。 有机分子中含氢基团(O— H, N— H, C— H)基频振动的倍频和合频都在此波段[2, 3], 通过扫描样品的近红外光谱, 可以得到样品中有机分子含氢基团的特征信息, 进而通过化学计量学的方法得到目标样品的物化参数。
为进一步了解和分析目标物分子激发态的光学性质, 理论上解释目标分子的近红外光谱产生机理, 许多研究团队都做了深入的研究工作[4, 5, 6, 7, 8, 9]。 如今, 随着密度泛函理论(density functional theory, DFT)[10]、 含时密度泛函理论(time-dependent density functional theory, TD-DFT)[11, 12]和计算机技术的不断发展和精确度的不断提高, 它们的应用范围也越来越广, 已经成为一种研究光谱产生机理非常重要的方法。 例如, Hadidi[13]等从电子结构理论模拟计算得到的光谱与实验结果非常吻合; Liu等对芳香族氨基酸的紫外和荧光光谱进行了理论研究和建模。 但对于化学战剂的近红外波段的振动特征峰的指认和产生机理分析较少有报道。
近红外光谱是检测化学战剂和危险化学品的常用方法, 但化学战剂和危险化学品对于实验环境要求高, 存在较大的危险性。 为更精细准确的研究化学战剂和危化品的近红外光谱, 利用DFT理论方法模拟战剂和危化品的近红外光谱可进一步支撑相关近红外光谱检测方法的应用与测试, 为化学战剂和危化品的实验补充空白。 本工作选取了沙林分子作为研究对象, 建立针对分子近红外光谱的模拟计算模型并验证该模型的可行性和准确性。 为在实际应用中更好的利用化学战剂近红外光谱提供借鉴。
在ChemSpider化学数据库中获取沙林分子的初始结构, 基于Gaussian16程序包, 通过密度泛函理论(DFT), 利用B3LYP/def2-SVP对沙林分子进行基态结构优化, 优化的收敛限度设定为标准, 各收敛限度为: 力的最大值(maximum force)< 0.000 45; 均方根(RMS force)< 0.000 3; 最大位移(Max. displacement)< 0.001 8; 均方根位移(RMS displacement)< 0.001 2。 达到各收敛限度后优化完成, 对优化完成后的基态结构在同一计算级别下进行简谐振动分析得到该结构下的简谐振动频率, 无虚频即证明优化完成的结构为分子的最稳定形态。 以优化后的沙林分子基态结构为计算对象进行非简谐振动分析, 计算理论选择广义振动二阶微扰理论(GVPT2)。 大量的研究证明, 用GVPT2计算此类有机体系非简谐振动有较高的精度和效率, 适合于近红外区域的研究。 通过该理论计算得到分子所有的非简谐振动模式、 频率以及强度, 根据倍频和合频振动绘制得到近红外光谱; 由于实验测定的近红外光谱范围为1 000~1 600 nm, 因此计算近红外范围内的非简谐振动频率时, 除了计算默认的分子二倍倍频和合频外, 还计算得到了沙林的三倍倍频和合频。
如图1所示, 通过DFT方法优化沙林分子的几何结构。 经对该优化后的结构进行振动分析, 未发现虚频, 表明该结构为沙林分子的基态最稳定结构。 其红外光谱基频振动模式如表1所示。
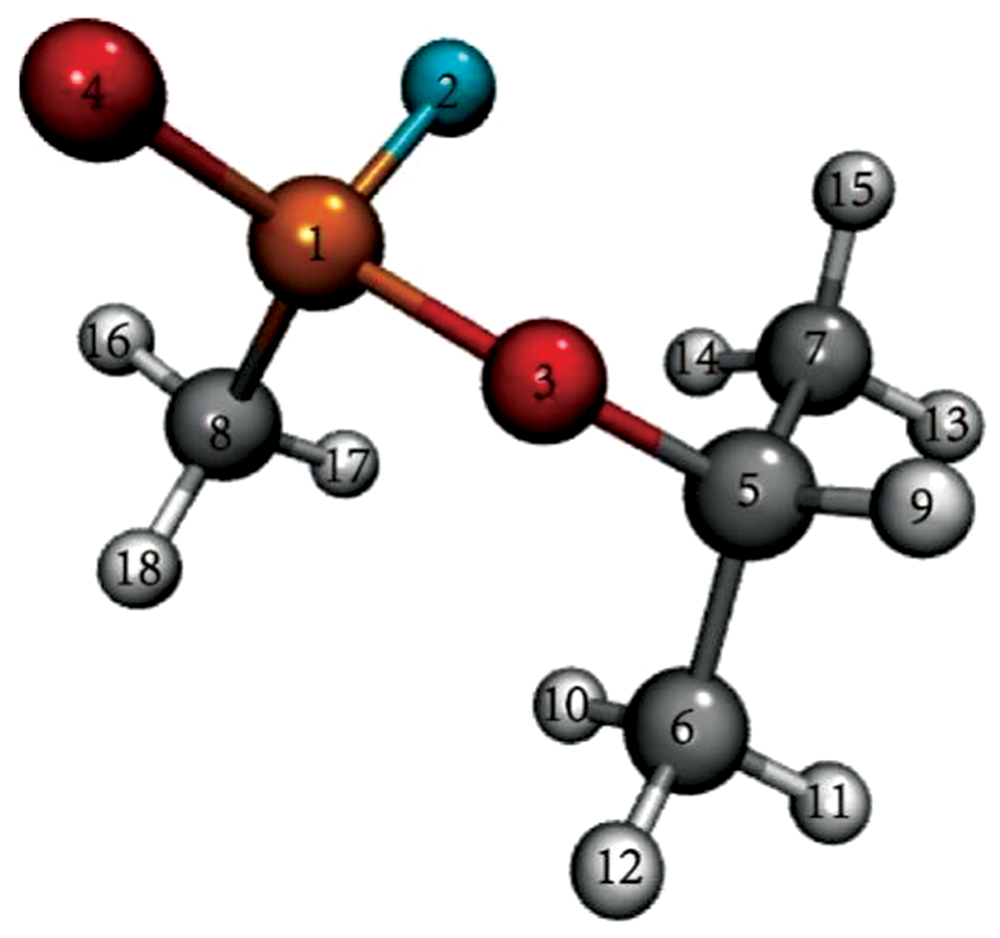 | 图1 沙林分子结构图 灰色为碳, 红色为氧, 橘色为磷, 蓝色为氟, 白色为氢Fig.1 The molecular structure of sarin gray represents carbon, red represents oxygen, orange represents phosphorus, blue represents fluorine, and white represents hydrogen |
| 表1 与近红外光谱相关的基频振动模式 Table 1 Fundamental vibration modes related to near-infrared spectroscopy |
分子能选择性吸收电磁波中红外区域的光, 从而引起分子振动。 该吸收特异性源于样品分子中的特征化学键。 因此红外光谱能提供分子的官能团和化学键的丰富结构信息。 一个化学键的振动通常会出现多个不同位置的吸收峰; 红外光谱上的某个峰可能是多个不同化学键的振动导致。 在中红外范围内, 吸收峰较多, 峰型复杂, 在检测化学战剂和危化品时, 存在图谱分析困难、 不精确等问题, 导致检测时间过长, 成本过高。
分子振动的非谐振性使分子从基态向高能级跃迁同时产生光谱。 相较于中红外范围, 吸收峰较少的近红外光谱能快速高效的定性分析大多有机样品, 能达到甄别和鉴别化学战剂的目的。
通过计算沙林分子的基频的振动模式, 中红外范围内共有49种振动模式, 导致中红外范围内峰型复杂, 其近红外范围内倍频、 合频仅有3个振动峰, 涉及的振动模式主要有18种(表1)。 选择900~1 700 nm作为研究的主要波段范围, 通过实验测定沙林分子的近红外光谱。 图2是沙林的实验近红外光谱, 存在2个明显的特征峰群, 一个在1 150 nm, 另一个在1 362 nm, 另外还有一个弱特征峰在1 500 nm。 计算模拟得到的近红外光谱同样存在三个振动峰, 和实验相符, 计算过程中光谱的半高全宽设定为20 cm-1。 由图2可以看出, 模拟得到的近红外光谱与实验结果吻合度较高, 该计算模型可以准确模拟得到化学战剂的近红外光谱, 通过进一步分析, 可以准确理解各吸收峰由哪几种倍频或合频产生。
 | 图2 沙林的近红外光谱 红色(左坐标轴): 计算光谱; 黑色(右坐标轴): 实验光谱Fig.2 The near-infrared spectra of Sarin red (left axis): calculated spectrum; black (right axis): experimental spectrum |
通过B3LYP/def2-SVP计算得到的近红外光谱可以用于分析实验光谱中吸收峰的振动模式。 在近红外范围内, 主要存在两个较为明显的振动峰, 分别位于1 150和1 362 nm, 1 500 nm也存在一个振动峰, 但1 500 nm的振动峰强度较低, 通过表2和表1分析上述特征峰是由哪些振动模式贡献造成的:
| 表2 沙林分子的理论近红外振动峰与主要振动模式 Table 2 Theoretical near-infrared vibration peaks and main vibration modes of sarin molecules |
首先分析1 150 nm峰的振动模式, 根据计算结果的分析, 该峰是由多个倍频和合频的组合振动贡献产生, 主要由分子中C— H伸缩振动的三倍倍频和分子中C— H原子非对称伸缩振动的三倍倍频以及对称及非对称伸缩振动基频的合频所贡献。 根据对1 150 nm红外振动峰中振动模式的指认, 强度较高的振动模式一般为某基频振动的三倍倍频贡献产生, 可以看出该峰主要是由于沙林分子中四个碳原子上的各种振动, 再由于倍频和合频组合所产生的红外振动峰, 可以看出近红外范围内主要的振动模式为C— H相关的振动且强度较高。
另一个主要特征峰位于1 362 nm, 该峰所包含的各个振动模式的红外强度相似, 只是位置有所差别, 因此整个红外振动峰较宽。 振动模式主要包括: 分子中C6, C7, C8原子上H原子的平面剪式运动的基频, C6, C7上H原子对称、 非对称伸缩运动基频的合频, C8原子上H原子平面剪式运动、 对称、 非对称伸缩振动基频的合频, 以及O3— P1键的对称和非对称伸缩振动、 H原子非平面摇摆和其对称、 非对称伸缩振动基频的合频。 根据对不同振动模式的指认, 可以看出, 强度较高的振动模式主要为分子中C上所连原子相关的振动, 强度较低的合频和倍频主要由其他的非C, H原子产生的振动产生。
最后分析1 500 nm峰位存在的一个由于强度过低被掩盖的红外振动峰, 该峰的振动模式主要有平面剪式运动、 整个分子的非平面扭转振动、 非对称伸缩振动基频的合频, 以及P— F键的伸缩振动。 由此可见, 1 500 nm附近的近红外振动峰主要由C8相关的振动模式贡献产生, 包含一定的非C, H原子的振动模式, 峰强度较低。
近红外范围内的振动光谱, 其中的振动峰主要是由于各基频振动的三倍倍频、 一个基频的两倍倍频和另一个基频相加组成的合频、 三个基频相加组成的合频, 这三种振动模式贡献导致。 其中与C, H相关的振动模式强度较高, 在波长较小的范围存在非C, H原子的振动模式, 如O— P, P— F等振动模式为沙林的特征振动模式, 在近红外范围内强度较低, 可以用理论计算模型得到精确的指认与分析。
采用DFT与TDDFT方法, 对目标化学战剂进行非简谐振动分析计算, 建立近红外光谱的理论计算模型。 计算其特征峰位并分析其产生原理, 与实验数据对比得到如下结论:
为了详细解释近红外光谱各吸收峰是由分子的何种振动模式的叠加产生, 通过DFT理论, 建立近红外光谱理论计算模型, 通过理论计算, 得到沙林分子在1 150, 1 362和1 500 nm处的三个特征峰。 通过实验测得沙林分子的近红外范围内的振动光谱, 存在2个明显的特征峰群, 一个在1 150 nm附近, 另一个在1 362 nm附近, 另外还有一个弱特征峰在1 500 nm附近。 可以看出理论计算结果与实验结果吻合度较高, 通过该计算模型可以用于模拟化学战剂和危化品的近红外光谱并分析得到各吸收峰是由其分子中对应原子振动的基频和倍频组合产生, 解释了其特征峰的产生机理, 达到鉴别和甄别化学战剂的目的。
后续研究拟将该模型应用于其余化学战剂和危化品的近红外光谱计算中, 对比计算光谱与实测光谱, 进一步验证该计算模型的准确性。 将计算数据整理成为数据库, 可为化学战剂和危险化学品的近红外光谱提供数据支撑。 填补某些化学战剂和危化品的空白。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|