

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
氟氯酰与正癸烷反应的瞬态光谱和机理探索研究
[闫华1  , 刘兴华
, 刘兴华2 , 丁勇3 , 赵志1 , 罗永锋1 , 武玉红1 , 颜澎1 , 董露1 , 王大喜4 ]
, 刘兴华|
|


作者简介: 闫 华, 女, 1974年生, 火箭军研究院硕博连读研究生, 副研究员 e-mail: hlary@163.com
氟氯酰(ClF3O)是一种极强的氟化剂和氧化剂, 极易与水和有机物发生爆炸性反应。 目前关于氟氯酰与水以及有机物等物质的反应机理不多见, 氟氯酰与水以及有机物等物质由反应物变成产物的过程有待研究。 采用ICCD瞬态光谱测量系统, 实时拍摄到无氧和有氧环境下氟氯酰和正癸烷反应的瞬态发射光谱; 采用量子化学理论方法对氟氯酰和正癸烷的反应机理进行了探索研究, 理论计算与试验结果相一致。 瞬态发射光谱试验结果表明, 在无氧环境下, 氟氯酰和正癸烷反应会产生CH和C2自由基, 证实了无氧时氟氯酰确实能与正癸烷发生反应, 显示出氟氯酰的高活性; 在有氧环境下, 则会产生OH, CH和C2自由基。 CH自由基强度最大的发射峰位于431.4nm, 归属于 A2 Δ-X2 П电子态之间的跃迁; C2自由基强度最大的发射峰位于516.3 nm, 归属于 A3 Пg- X3 Пu电子跃迁; OH自由基强度最大的发射峰位于309.5 nm, 归属于 A2 Σ+ -X2 Пi电子跃迁。 量子化学理论计算结果表明, ClF3O与正癸烷的反应始于ClF3O中具有较多负电荷的F原子向正癸烷分子中间的H原子进攻生成HF, 该引发反应活化能很低, 并大量放热。 在无氧环境下, 氟氯酰与正癸烷可能发生氟代反应, 反应产物为ClFO、 HF和相应的氟代烷烃等。 氟代烷烃可能会发生脱氢反应生成C10H20F, 接着裂解为C4H9及氟代烯烃C6H11F; C4H9进一步分解为C2H5和C2H4, 最终形成CH和C2自由基等。 有氧环境下反应初始步骤与无氧条件下相同, 当反应进行到一定程度, 产生烷烃自由基之后, O2参与反应, 形成过氧自由基, 过氧自由基继续分解, 产生OH, CH和C2自由基。 在氧气参与下, 反应过程中产生大量的OH自由基, 加速反应的进程, 宏观上表现为正癸烷被引发爆燃与燃烧。 这些自由基和中间体对于揭示氟氯酰和正癸烷反应的微观机理具有重要的指示意义。 氟氯酰和正癸烷的反应机理与试验结果均证实: 小自由基CH、 OH和C2是氟氯酰与正癸烷反应过程中的重要中间产物, 这对于认识氟氯酰与正癸烷反应的微观过程非常重要, 也为氟氯酰的武器化应用奠定了一定的理论基础。
, LIU Xing-huaChlorine trifluoride oxide (ClF3O) has stronger corrosive and oxidizing properties than other chlorine fluorides such as ClF3. It can react with numerous materials, e.g., water and hydrocarbons. The reaction between ClF3O and organic hydrocarbons may occur at quite a low temperatures and cause an explosion. So far, however, no detailed information about the reactions is available. Using an intensified charge-coupled device (ICCD) system, transient emission spectra of the reaction of ClF3O and n-decane were measured in a spectral range of 200~850 nm. Using density functional theory (DFT) method were performed to investigate the reaction mechanism of ClF3O and n-decane. All calculated results are consistent with the experimental data, which indicates that the present results are credible. The emission spectra of CH and C2 radical intermediates were observed in the reactions of ClF3O and n-decane under a no-oxygen environment, and this shows that ClF3O is a highly reactive compound. The detection of the CH, C2 and OH radical intermediates shows clearly that a large amount of energy was released during the reaction between ClF3O and n-decane under an oxygen environment. The primary peak was found at 431 nm corresponding to the A2 Δ-X2 Π electronic transition of the CH radical. The peak at 516 nm produced by the A3 Пg- X3 Пu electronic transition of the C2 radical was also observed. The peak at 309 nm corresponds to the A2 Σ+ -X2 Пi electronic transition of the OH radical was also found. The results of the calculations showed that the F atom on ClF3O attacks the H atom on n-decane to initialize the reactions, and a F atom on ClF3O abstracted h atom on n-decane to produce HF. The initial reactions were considered to be barrier-less reactions and extremely exothermic. Under a no-oxygen environment, a fluorination reaction occurred between ClF3O and n-decane, and the products were ClFO, HF and corresponding fluoroalkanes. Fluoroalkanes may undergo dehydrogenation to form C10H20F. Then it is cleaved into C4H9 and C6H11F, then C4H9 further decomposed into C2H5 and C2H4, and finally formed CH and C2 radical. The initial steps of reaction in the aerobic environment were the same as in an anaerobic condition. When the reaction proceeded to a certain degree, after producing alkane radicals, O2 formed peroxic radicals, and peroxic radicals continue to decompose to form CH, C2 and OH radical intermediates. In the presence of oxygen, many OH radicals were produced in the reaction process, which accelerated the process of reaction. Macroscopically, n-decane was initiated by deflagration and combustion. Results show that the main emission bands are attributed to OH, CH and C2 radicals produced during the reaction process of ClF3O and n-decane, which reveals that small OH, CH and C2 radicals are important intermediate products in the reaction process of ClF3O and n-decane. This is very important for understanding the micro-process of reaction of ClF3O and n-decane. It also play an important theoretical foundation for the application of the weapon of ClF3O.
氟氯酰(ClF3O)是一种极强的氟化剂和氧化剂, 其化学活性要比已知的强氧化剂ClF3、 ClF5还要活泼, 极易与水和有机物发生爆炸性反应。 文献[1, 2, 3, 4]研究了ClF3与H2O、 C3H6O和CH4的反应机理, 但是关于氟氯酰与水以及有机物等反应机理并不多见, 氟氯酰与水以及有机物等由反应物变成产物的过程有待研究。 正癸烷(n-Decane, 煤油的模型化合物)是非常重要的单组分碳氢燃料, 是商用和军用煤油的重要组分, 也是煤油替代燃料常用的主要组分。
利用高空间分辨率和高灵敏度的各种快速响应光谱技术, 从微观角度研究物质的物性变化在国内外已经成为一种新的发展趋势[5, 6, 7, 8, 9]。 瞬态光谱测量技术在研究自由基方面有着独特的优势, 可以在燃料燃烧反应实验中, 一次性地记录特征谱线(即波长与光强对应关系), 获得反应中间自由基的高分辨特征发射光谱。 这些中间自由基信息为从分子水平上认识氟氯酰和正癸烷反应的微观过程提供了第一手实验依据。
本研究采用瞬态光谱测试系统对氟氯酰与正癸烷反应过程进行了实时光谱测量, 拍摄到在无氧和有氧环境下, 氟氯酰和正癸烷反应中间自由基的发射光谱。 采用量子化学理论方法, 对氟氯酰与正癸烷的反应机理进行了探索研究, 对可能发生的基元反应通道进行数值仿真, 揭示氟氯酰与正癸烷反应的微观机理, 理论与试验结果相一致。
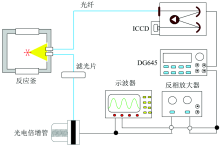
ICCD(增强电荷耦合器件)相机是像增强器(image intensifier)与电荷耦合器件(charge coupled device, CCD)相组合的光学测试仪器。 其测试原理是像增强器获得燃烧火焰的光学信号后输出550 nm绿光, 经中继光学元件与可见光CCD耦合, CCD把光敏元上的光信息转换成与光强成比例的电荷量。
ICCD瞬态光谱仪测试系统分三个部分: 第一部分是可视反应釜, 可进行无氧和有氧条件下的化学反应; 第二部分是同步触发系统, 以CH自由基的出现作为反应起点[10, 11], 控制ICCD同步触发; 第三部分是光谱采集系统, 由光谱仪和ICCD组成。
图1为瞬态光谱测量系统。 在可视反应釜中, 反应物正癸烷(3.0 g)与氟氯酰(0.5 g)发生化学反应并发出光信号。 在反应区域正上方连接两条光纤, 一条光纤直接与ICCD连接, 另一条光纤与光电倍增管(PMT)相连。 反应发出的光经滤光片得到431 nm的光信号, 然后由PMT转变为电信号(~-50 mV), 再经反相放大器继续放大, 达到触发DG645的电压(5 V)后, DG645触发光谱仪和ICCD, 实现发射光谱的同步检测。 光谱采集波长为250~800 nm, 光栅规格为1 200 g· mm-1, 曝光时间为100 ms。 每次实验前, 光谱仪均通过汞灯进行标定。
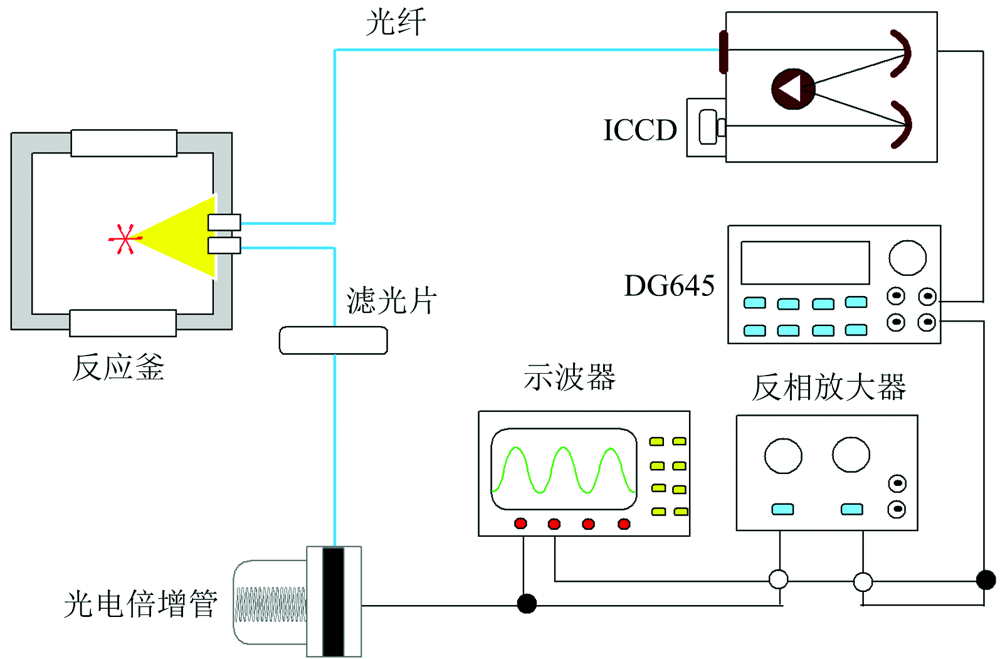 | 图1 瞬态光谱测量系统Fig.1 Transient spectral measurement system |
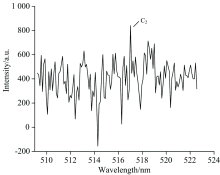
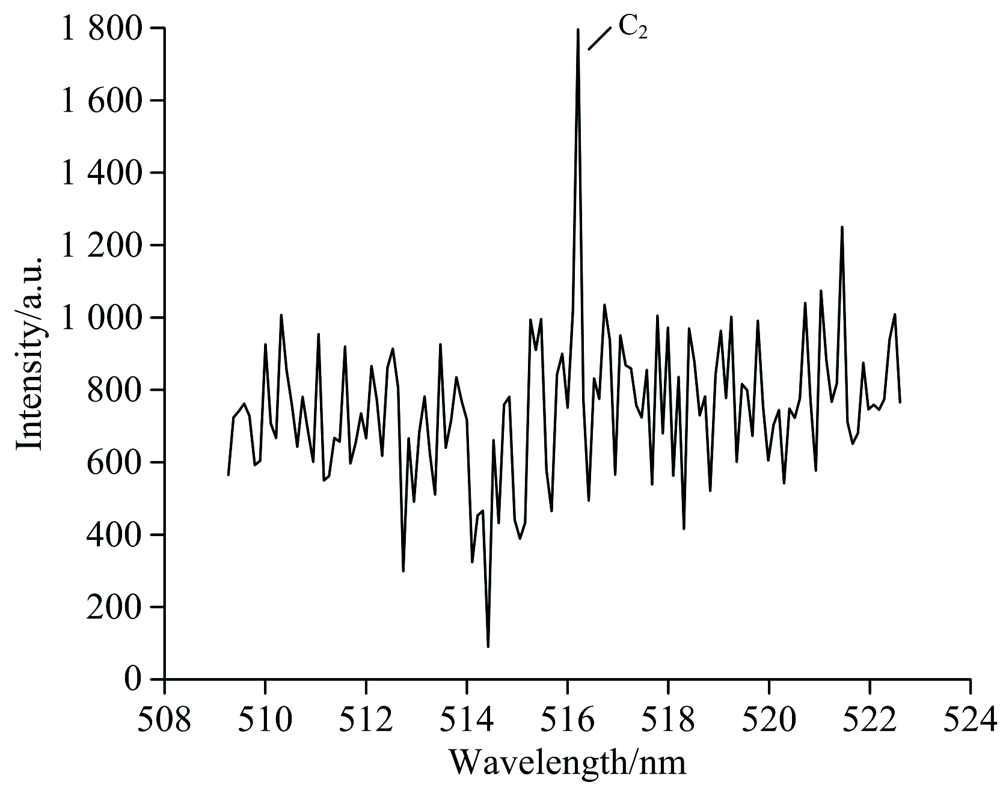
拍摄得到氟氯酰和正癸烷反应产生的CH和C2高分辨特征发射光谱, 分别如图2和图3所示。 光谱图中, 横轴为波长(单位为nm), 纵轴为光强。 选择CH Swan带系的431.4 nm谱线[5, 6, 7], C2的516.5 nm谱线。
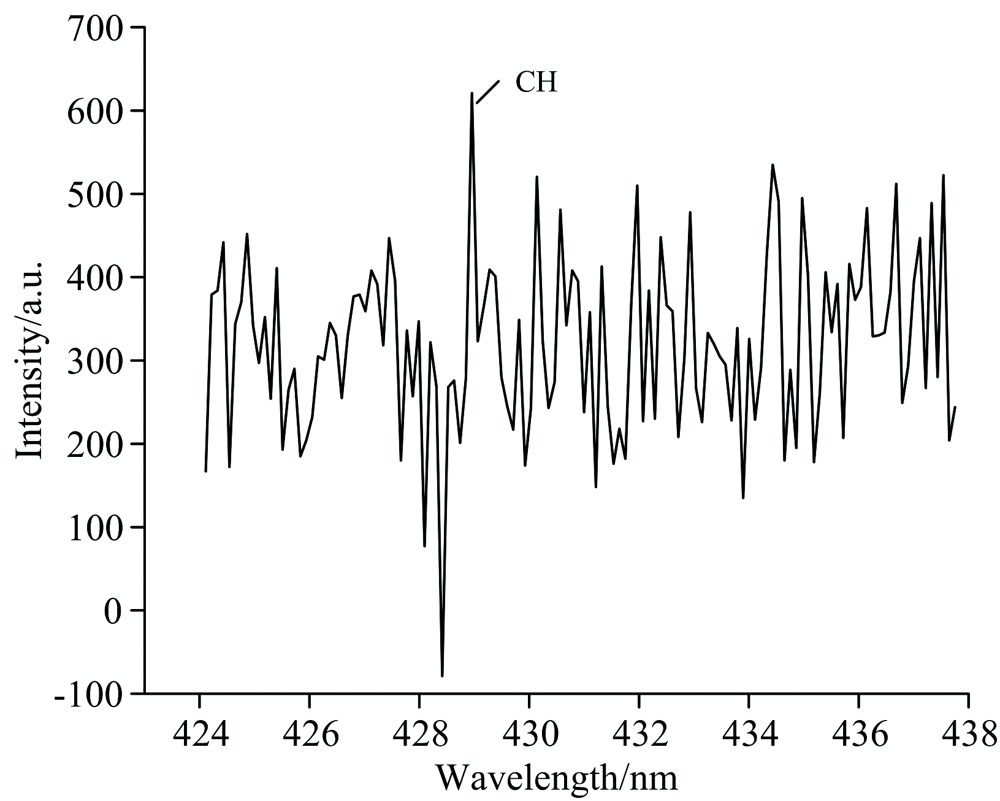 | 图2 无氧时氟氯酰和正癸烷反应产生的CH发射光谱Fig.2 The CH emission spectrum produced by the reaction of ClF3O and n-decane in the absence of oxygen |
 | 图3 无氧时氟氯酰和正癸烷反应产生的C2发射光谱Fig.3 The C2 emission spectrum produced by the reaction of ClF3O and n-decane in the absence of oxygen |
通过对各光谱带的分析和指认, 确定其归属于CH和C2自由基的特征光辐射。 由此可知, 在无氧环境下氟氯酰能引发正癸烷发生反应, 导致正癸烷裂解, C— C和C— H键发生断裂产生CH和C2自由基。 从实验上证实了无氧时氟氯酰确实能引发正癸烷发生反应, 显示出了氟氯酰的高活性。
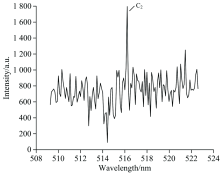
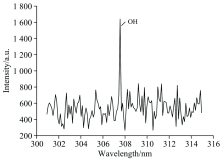
拍摄得到了有氧时氟氯酰与正癸烷反应产生的CH, OH和C2高分辨发射光谱, 分别如图4、 图5和图6所示。
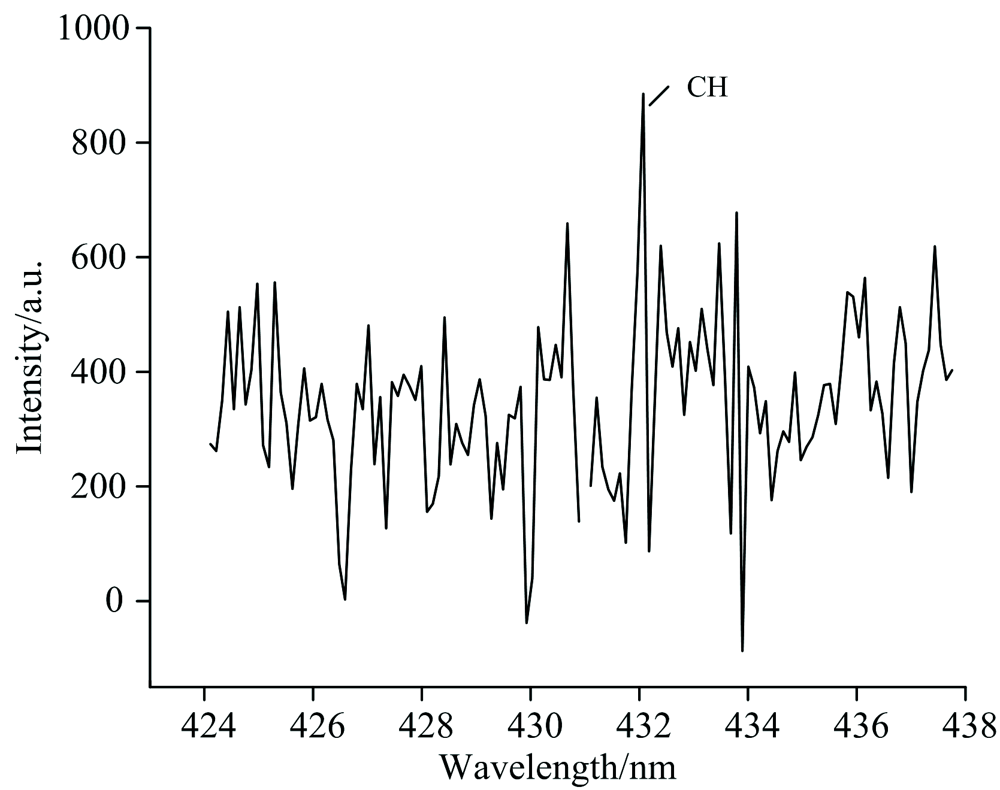 | 图4 有氧时氟氯酰和正癸烷反应过程中CH发射光谱Fig.4 The CH emission spectrum produced by the reaction of ClF3O and n-decane in the presence of oxygen |
 | 图5 有氧时氟氯酰和正癸烷反应过程中OH发射光谱Fig.5 The OH emission spectrum produced by the reaction of ClF3O and n-decane in the presence of oxygen |
 | 图6 有氧时氟氯酰和正癸烷反应过程中C2发射光谱Fig.6 The C2 emission spectrum produced by the reaction of ClF3O and n-decane in the aerobics |
在200~850 nm, 检测到CH, C2和OH自由基, 其中CH (0, 0)带的带头波长为431.4 nm, OH (0, 0)带的带头波长为306.4 nm, C2 (0, 0)带的带头波长为516.5 nm, 显示有较强的CH, OH和C2自由基发射光谱带, 可知这些自由基含量比较高, 表明在氧气参与下, 氟氯酰与正癸烷发生剧烈反应, 释放出大量的热量。
各发射峰归属及类型总结于表1。 C2自由基的发射峰均归属于A3П g-X3П u电子跃迁, 其中强度最大的发射峰波长516.3 nm; CH自由基的发射峰分为三种类型, 分别为C2Σ --X2П , B2Σ --X2П 和A2Δ -X2П 电子态之间的跃迁, 其中强度最大的发射峰波长431.4 nm; OH自由基的发射峰均归属于A2Σ +-X2П i电子跃迁, 其中强度最大的发射峰波长309.5 nm。
| 表1 氟化物和正癸烷反应的发射峰归属 Table 1 Emission peak attribution of the reaction of ClF3O and n-decane |
采用密度泛函理论, 在B3PW91/6-31++G(d)水平上, 对氟氯酰与正癸烷的反应机理进行了探索研究, 分析了氟氯酰与正癸烷反应的微观过程。
2.3.1 无氧时氟氯酰和正癸烷反应机理
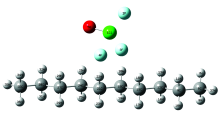
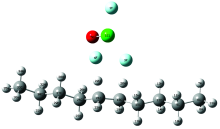
采用密度泛函理论方法B3PW91/6-31++G(d)对ClF3O与正癸烷在无氧条件下的反应过程进行了计算。 图7是氟氯酰与正癸烷的初始作用模型; 图8是氟氯酰与正癸烷第一步引发反应的过渡态; 表2是无氧时氟氯酰与正癸烷反应的热效应计算结果。
 | 图7 氟氯酰与正癸烷初始作用模型Fig.7 The initial action model of ClF3O and n-decane |
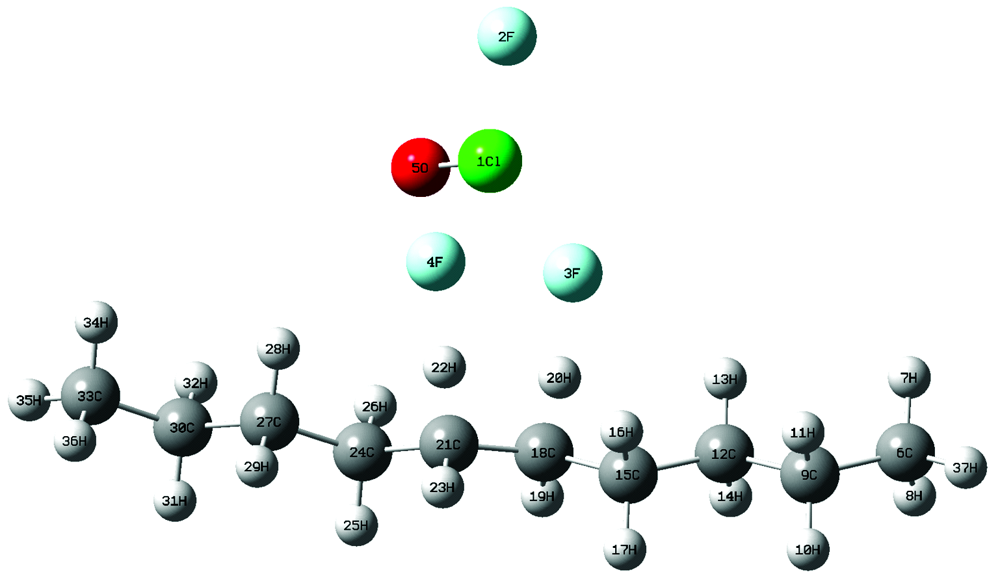 | 图8 氟氯酰与正癸烷第一步反应的过渡态Fig.8 The transition state of the first step reaction of ClF3O and n-decane |
| 表2 无氧时氟氯酰与正癸烷反应的热效应计算 Table 2 The thermal effect of the reaction of ClF3O and n-decane in the absence of Oxygen |
从图7和图8中可以看出, 过渡态结构与反应始态结构相似, 表明该类反应为强放热反应。 表2中ClF3O与正癸烷引发步放热量为-309.24 kJ· mol-1, 证实第一步反应大量放热, 表明了氟氯酰的高活性。 计算结果表明: ClF3O与正癸烷的反应始于ClF3O中具有较多负电荷的F原子(3F和4F)向正癸烷分子中间的H原子(20H和22H)进攻生成HF, 这一步是整个反应的引发步骤活化能很低, 并且大量放热, 这些都预示着该反应具有强烈自发进行的趋势。
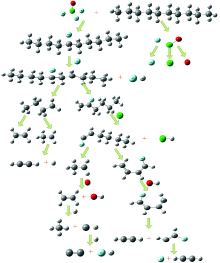
图9为无氧时氟氯酰引发正癸烷可能的反应机理。 ClF3O易于C— H键发生取代反应, 氟氯酰与正癸烷发生氟代反应, 反应产物为ClFO、 HF和相应的氟代烷烃等。 氟代烷烃可能会发生脱氢反应生成C10H20F, 然后裂解为C4H9及氟代烯烃C6H11F, C4H9进一步分解为C2H5和C2H4, C6H11F分解为C2H5和C4H5F, C2H4会与H反应生成CH4和CH等自由基和小分子, CH与F反应生成C2自由基等。
 | 图9 无氧时氟氯酰引发正癸烷的反应机理Fig.9 The reaction mechanism of ClF3O and n-decane in the absence of oxygen |
2.3.2 有氧时氟氯酰与正癸烷反应机理探索
有氧条件下氟氯酰与正癸烷的反应, 初始步骤与无氧条件下相同, 当反应进行到一定程度, 产生烷烃自由基之后, O2参与反应, 进行后续步骤。
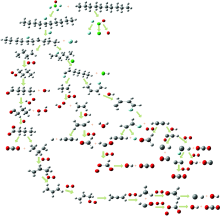
图10为有氧时氟氯酰引发正癸烷反应的可能反应机理, 表3是有氧时氟氯酰与正癸烷反应的热效应计算结果。 在引发阶段, 氟氯酰首先攻击正癸烷分子上的H, 与正癸烷发生氟代反应, 氟代烷烃脱氢和裂解生成烷烃自由基等, O2参与反应, 形成过氧自由基, 过氧自由基分解会生成醛类、 酮类、 OH和CO2等, 再继续发生脱氢裂解反应生成C2H5和C2H4等小分子和C2H3, CH3, C2H, CH和C2等自由基, 最后由小分子和自由基的氧化反应生成最终产物。 从图10和表3可以看出, C2H+O→ CO+CH和C2H+O2→ CO2+CH这两个基元反应放热量较大, 说明这两个通道可能是CH的主要生成机理, 与相关研究预测的CH主要生成机理相一致。 反应2CH+2F→ 2HF+C2放热量也较大, 说明此反应可能是氟氯酰引发正癸烷反应产生C2自由基的主要生成机理。 OH涉及多个基元反应, 作为反应物和产物不停地出现和消失, 与相关研究预测的OH可能涉及多步基元反应相一致。 在氧气参与下, 反应过程中会产生大量的OH自由基, 加速反应的进程, 宏观上表现为正癸烷被引发爆燃与燃烧。 这些自由基对于揭示氟氯酰和正癸烷反应的微观机理具有重要的指示意义。
 | 图10 有氧时氟氯酰引发正癸烷的反应机理Fig.10 The reaction mechanism of ClF3O and n-decane in the presence of oxygen |
| 表3 有氧时氟氯酰与正癸烷反应的热效应计算 Table 3 The thermal effect of the reaction of ClF3O and n-decane in the presence of Oxygen |
综上所述, 氟氯酰和正癸烷的反应始于ClF3O中具有较多负电荷的F原子向正癸烷分子中的H原子进攻生成HF, 该引发步骤活化能很低, 并且大量放热。 在无氧环境下, 氟氯酰与正癸烷反应产生CH和C2自由基, 在有氧环境下, 则产生OH, CH和C2自由基。 这些结论与拍摄到的氟氯酰和正癸烷反应过程OH, CH和C2的高分辨特征发射光谱结果相一致, 证实了量子化学理论计算的正确性。
通过瞬态光谱测量试验方法, 获得了氟氯酰与正癸烷反应过程中的CH, OH和C2高分辨特征发射光谱。 采用量子化学理论方法, 研究了氟氯酰和正癸烷的反应机理, 结果表明无氧时二者反应会产生CH和C2自由基, 有氧时会产生OH, CH和C2自由基。 理论与试验结果相一致。
理论与试验结果均证实: 小自由基CH, OH和C2是氟氯酰与正癸烷反应过程中的重要中间产物, 对于认识氟氯酰与正癸烷反应的微观过程非常重要, 也为氟氯酰的武器化应用奠定了一定的理论基础。
致谢: 四川大学李象远教授、 李萍教授和清华大学徐胜利教授对本工作的支持和帮助!
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|