

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
黄腐酸络合铜离子光谱学特征及机理构建
[徐恒山2  , 巩冠群
, 巩冠群1, 2, * , 张英杰1, 2 , 袁飞2 , 张永霞2 ]
, 巩冠群, 张英杰|
|


作者简介: 徐恒山, 1997年生, 中国矿业大学化工学院硕士研究生 e-mail: 18821671546@163.com
二价金属离子Cu2+在很多工矿企业周围水源及土壤中存量超标, 造成生态环境恶化, 传统的药剂及生物处理容易产生二次污染。 黄腐酸由性质相似分子团簇构成, 具有水溶性好、 络合作用强及化学活性高的特点, 对环境中Cu2+分布、 迁移和生物利用度可以实现高效控制与环保处理, 是近年科学研究热点。 现代多光谱表征分析有助于揭示黄腐酸与金属离子作用过程构效关系变化、 环境效应及重金属离子迁移行为规律, 对黄腐酸与Cu2+络合过程特点及作用机理研究具有重要科学价值。 综述了近年来黄腐酸与Cu2+络合作用相关基础理论研究, 通过红外光谱、 三维荧光光谱及差分光谱等方法对黄腐酸与Cu2+络合前后表征对比分析和学科交叉协同研究, 探讨了pH、 离子浓度以及黄腐酸组分构成差异等对络合过程的影响, 揭示了黄腐酸与Cu2+络合作用位点的结构特性及作用规律, 羧基与酚羟基等含氧酸性官能团是Cu2+与黄腐酸络合的主要位点, 羧基型位点络合Cu2+作用显著, 酚羟基型位点有助于增加Cu2+络合物稳定性, 含氮官能团也在络合过程中发挥重要作用。 在此基础上, 进一步指出pH值的变化将改变黄腐酸活性位点对Cu2+的亲和力, 原因主要与活性位点上Cu2+与H+之间的离子交换以及黄腐酸的静电吸引力有关; 不同结构特征的黄腐酸对Cu2+络合效果主要体现在具有不同数量的酚羟基、 羧基以及含氮官能团活性位点; 溶液中共存Fe3+, Mg2+和Al3+等离子会与Cu2+在黄腐酸的活性结合位点上产生显著络合竞争; 同时溶液环境中K+和Na+等非强吸附作用离子浓度增加, 使得溶液中大量正电荷离子就近进入黄腐酸的电子层而增强电荷屏蔽效应, 进而也抑制Cu2+与黄腐酸络合。 总结并展望了黄腐酸相关学科技术理论在现代农业、 生态修复及环境治理等领域科学应用共存的问题及挑战。
The divalent metal ion Cu2+ exceeds the standard in water sources and soils around many industrial and mining enterprises, causing deterioration of the ecological environment, and traditional chemical and biological treatments are prone to secondary pollution. Fulvic acid is composed of molecular clusters with similar properties. It has the characteristics of good water solubility, strong complexation and high chemical activity. It can efficiently control the distribution, migration and bioavailability of Cu2+ in the environment and is a hot spot in scientific research in recent years. Modern multispectral characterization analysis is helpful to reveal the changes in the structure-activity relationship between fulvic acid and metal ions, environmental effects and the migration behavior of heavy metal ions. It has important scientific value for studying the characteristics and mechanism of the complexation process of fulvic acid and Cu2+. This article reviews the basic theoretical research on the complexation of fulvic acid with Cu2+ in recent years. This paper further analyzes the characterization of fulvic acid and Cu2+ before and after complexation through infrared spectroscopy, fluorescence spectroscopy, differential spectroscopy, and interdisciplinary collaborative research. The effects of pH, ion concentration and the difference in composition of fulvic acid on the complexation process were discussed. The complex sites’ structural characteristics and action rules between fulvic acid and Cu2+ are revealed. Oxygen-containing acidic functional groups, such as carboxyl and phenolic hydroxyl, are the main complex sites between the complexation process of Cu2+ and fulvic acid. The carboxyl site has a significant ability to complex Cu2+. The phenolic hydroxyl site is helpful to increase the stability of the Cu2+and fulvic acid complex, and the nitrogen-containing functional group also plays an important role in the complex process. On this basis, this article further points out that the change of pH value will change the affinity of the active site of fulvic acid to Cu2+, the reason is mainly related to the ion exchange between Cu2+ and H+ on the active site and the electrostatic attraction of fulvic acid. The difference in FA components affects the complexation of FA and Cu2+, which is mainly reflected in the different numbers of phenolic hydroxyl, carboxyl and nitrogen-containing functional groups in different FA. The coexistence of Fe3+, Mg2+ and Al3+ the solution will have significant competition with Cu2+ at the active binding site of fulvic acid. At the same time, the concentration of non-strong adsorption ions such as K+ and Na+ in the solution environment increases, so that a large number of positively charged ions in the solution enter the electronic layer of fulvic acid nearby to enhance the charge shielding effect, inhibiting the complexation of Cu2+ and FA. Finally, this paper summarizes and looks forward to the problems and challenges of coexistence of scientific application of fulvic acid-related disciplines and technical theories in modern agriculture, ecological restoration and environmental governance.
腐殖质(humus, HS)对金属离子的化学迁移、 环境浓度分布、 微生物利用与吸收[1]有重要影响。 根据HS在不同pH环境下溶解特性, HS分为可溶于酸碱的黄腐酸(fulvic acid, FA)、 可溶于碱但不溶于酸的腐植酸(humic acid, HA)以及既不溶于酸也不溶于碱的腐黑物[2, 3, 4]。 HA含大量官能团, 分子尺度从几纳米到100 nm; FA分子半径约为1 nm, 分子结构中含有大约5~10个可电离的羧基和最多一个含N或S官能团, FA与金属离子络合物比HA的更稳定[5]; 同时, 由于具有较多羟基、 羧基等亲水基团, 相较于HA络合物, FA更具有亲水性, 水溶液中分散性更好[6]。 因此, FA通过络合作用对水体、 土壤中不同金属离子浓度控制、 迁移规律及转化利用研究具有重要价值[7], 是近年科学研究热点。
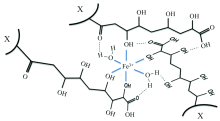
FA与金属离子相互作用的位点是特定的, 不同金属离子与FA相互作用的位点也不尽相同[8]。 FA来源物质和生成途径的多样性, 导致FA分子量、 构象、 质子亲和力以及羧基、 酚羟基等含氧官能团数量存在一定差异[9]。 FA分子不同特性构成影响金属离子结合过程及作用规律, 与金属离子的键合主要与羟基和羧基有关, 而形成FA络合物分子特性不仅与FA的分子结构有关, 还与其分子中官能团数量、 种类和构象相关, 且后者还可以通过金属配位络合物的形成来构成超分子结构[10](图1)。 目前, FA与金属离子的相互作用机理还缺少精准、 全面、 系统性研究。
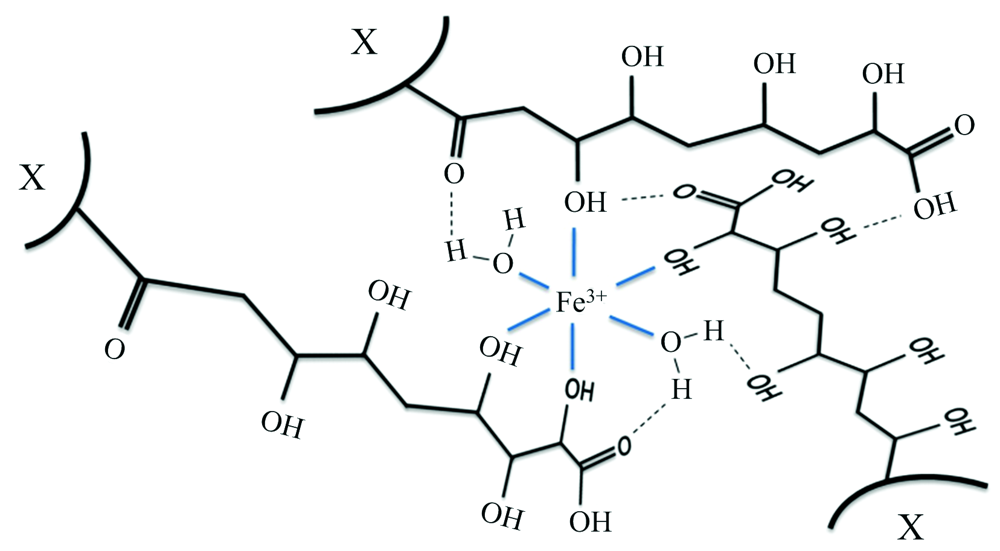 | 图1 铁离子与FA四种羟基配位络合作用[10] * X表示另一个大分子结构Fig.1 The coordination complexation of iron ions with the four hydroxyl groups of FA[10] |
铜是生物体物质代谢和生化反应过程不可缺少的微量元素, 主要以离子的形式被生物吸收转化利用, 在水溶液中以一价亚铜离子(Cu+)和二价铜离子(Cu2+)两种氧化态存在, 铜在两种氧化态之间的转化影响其反应性、 溶解性和生物利用度。 二价金属铜离子Cu2+在很多工矿企业周围水源及土壤中存量超标, 对生态环境造成了重要影响, 传统的药剂及生物处理容易产生二次污染, 寻找绿色环保、 性能优良的处理方法是目前亟待解决的重要科学问题。 研究表明, Cu2+与FA主要以络合的形式存在水环境中, 不能立即被生物利用, 但是能有效降低铜毒性[11, 12, 13]。 研究FA与Cu2+的络合作用机理有助于揭示环境中铜的具体形态、 毒性控制和生物利用度等, 进而实现FA与Cu2+络合规律及机理的科学应用。 本工作结合红外光谱、 三维荧光光谱及差分光谱等多光谱谱图特征表征研究结果, 重点阐述FA与Cu2+的络合过程规律、 作用机理及影响因素, 并指出未来发展的问题和挑战。
目前, FA与Cu2+作用规律研究还不是非常深入, 尤其是对其作用过程机理的揭示尚无科学定论。 有研究[14, 15]认为FA与Cu2+之间的相互作用为络合反应, 由特定的络合位点控制, 其中, 羧基与酚羟基等含氧酸性官能团是Cu2+与FA络合的主要位点, 但FA的含氮官能团对络合过程也有一定的影响, 芳香碳结构和疏水性结构也可能有助于FA与Cu2+的络合。 近年来, 研究者通过傅里叶变换红外光谱(Fourier transform infrared spectrometer, FTIR)、 三维荧光光谱(three dimensional fluorescence spectrum, 3DEEM)等对Cu2+与FA络合前后进行交叉表征, 对Cu2+与FA络合前后的差异进行解释和机理阐述, 有力促进了相关研究进程。
Ephraim 等[16, 17]发现聚电解质效应和功能异质性对FA与Cu2+络合平衡有影响, FA水溶液中分子组分比例依次为: pK值为1.8的羧酸占24.5%, pK值为3.4的羧酸占30.4%, pK值为4.2的羧酸占据22.4%, pK值为5.7的酸性醇(烯醇)占据22.7%; 与Cu2+与FA络合形成双齿结构有关的是pK值为3.4的羧基和pK值为5.7的羟基, 而且FA分子在低pH值下氨基羧酸位点相对较少, 对低浓度Cu2+可以实现高度选择性络合, pH值在3.5~6的实验结果与计算模型预测结果相符; 但是, 当pH值超过6时, FA与Cu2+络合特性预测与实验不符。 这可能与溶液中Cu2+在 pH> 5时以[Cu(OH)]+的形式存在, 而[Cu(OH)]+又可能在失去质子的同时, 与FA-Cu2+络合物发生不可逆的相互作用有关。 虽然该研究发现FA与Cu2+络合位点, 但未能对FA与Cu2+络合位点(羧基与酚羟基)进行详细解释, 作用机理仍需深入研究。
不同浓度下, Cu2+与FA羧基和酚羟基结合, 表现出明显的双齿络合特征, 作用过程部分非特异性静电相互作用一般较小可忽略不计[18, 19]。 与FA作用过程中Cu2+分为可溶性Cu2+、 与羧基类位点络合的Cu2+、 与酚羟基类位点络合的Cu2+和位于Donnan相中的Cu2+; 而且与酚羟基类位点络合的Cu2+数量比羧基类位点络合Cu2+数量少1~2个数量级[18, 19, 20, 21]。 因此, FA与Cu2+的络合数量主要受羧基类位点控制, 酚基型位点的作用较小, 但酚羟基类位点有助于FA与Cu2+的络合物的稳定性, 羧基和酚羟基为FA络合Cu2+过程主要络合作用位点[20, 21]。 目前, FA分子结构中其他官能团在Cu2+络合过程中是否有作用及作用机理还需理论及实验数据支撑。
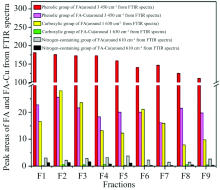
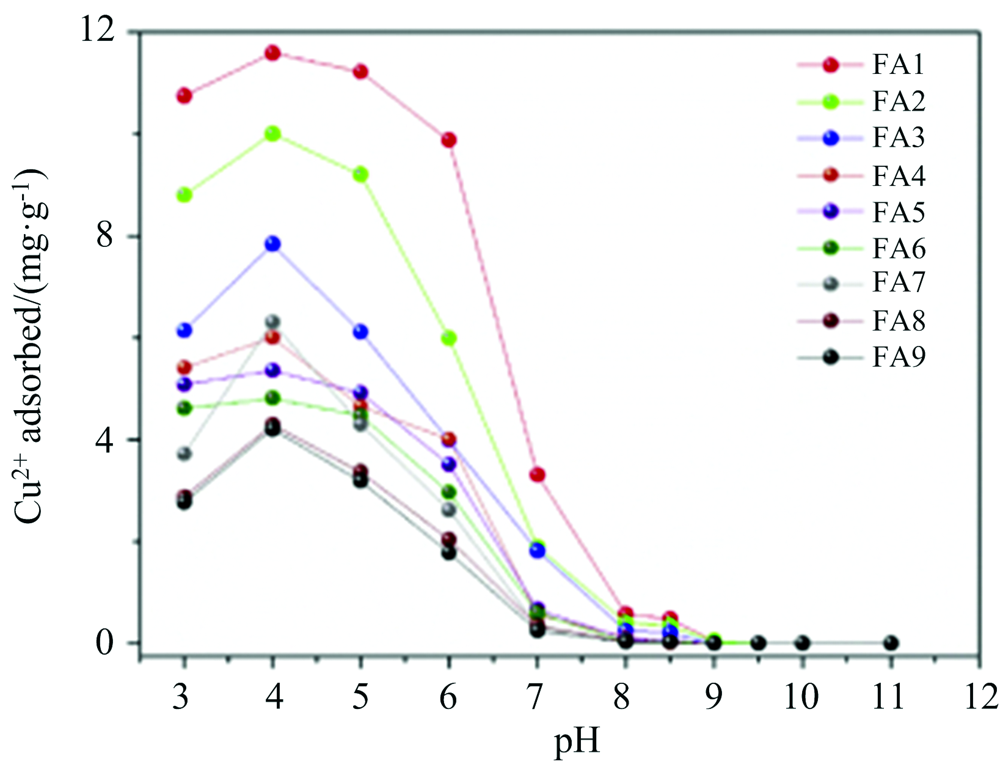
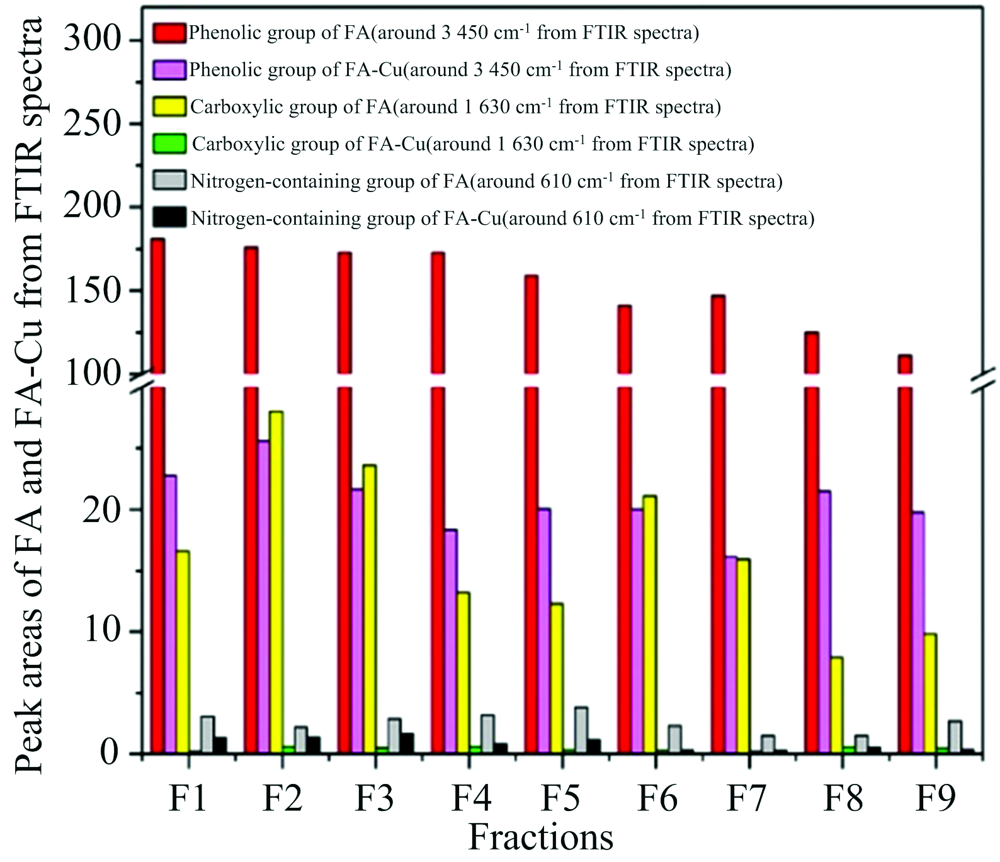
利用FTIR及3DEEM光谱研究发现[18], Cu2+在FA上的吸附主要有离子交换和表面络合两种活性位点, 表面络合位对Cu2+的吸附亲和性高于离子交换位, 表面络合位的吸附容量小于离子交换位的吸附容量, Cu2+在FA上的吸附主要以表面络合为主。 同时, 对FA与Cu2+总吸附容量和最大吸附容量研究也表明羧基和酚羟基为FA与Cu2+络合的主要络合位点[23, 24]。 通过比较Cu2+结合前后FA与FA-Cu络合物的FTIR特征官能团光谱差异[22](见图2), 发现官能团-OH峰强度在3 447 cm-1附近发生了急剧下降(大约偏移了20 cm-1), -COOH峰强度在1 630 cm-1附近发生了急剧下降(其中位移约20 cm-1), 证实两种基团已参与络合反应; FA-Cu络合物峰强度在1 120(偏移约5 cm-1)和1 035 cm-1(偏移约10 cm-1)之间减小, 表明FA-Cu络合物是通过对羟基中C-O的高亲和力而形成; 610 cm-1附近的峰强度(几乎没有偏移发生)降低, 表明含氮基团可能部分参与了吸附过程; FA-Cu络合物在1 370~1 400 cm-1附近峰强度增加, 意味着吸附后COO-含量增加[19, 20, 21, 22]。
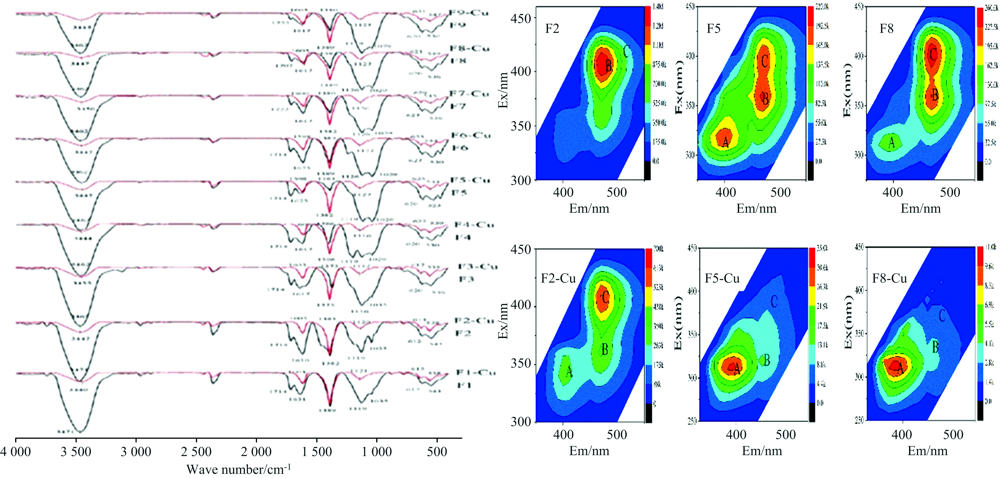 | 图2 9种FA络合Cu2+前后FTIR及3DEEM谱图[22]Fig.2 FTIR and 3DEEM spectra of 9 kinds of FA before and after complexing Cu2+ [22] |
FA与Cu2+络合前后3DEEM光谱显示(A峰和B峰均与高度共轭基团相关(如醌基), C峰与羧基和羰基相关, 在Cu2+吸附后, B和C峰均发生荧光猝灭, 表明吸附过程包括羧基的结合作用, 峰A的荧光强度增加表明FA-Cu络合物可能比FA包含更多的CO-结构, 与FTIR光谱一致, 也证实了羧基和酚羟基为FA与Cu2+络合的主要位点[20, 21, 22, 23, 24]。 FA分子结构中羧基、 酚羟基、 含氮基团等含量对Cu2+络合起到决定作用(图2)[22], 但是该研究基于离子交换和表面络合的两种吸附位点的假设, 没有综合考虑FA分子结构的整体效应, 也造成了该理论假设的部分缺陷。
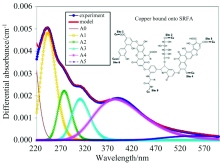
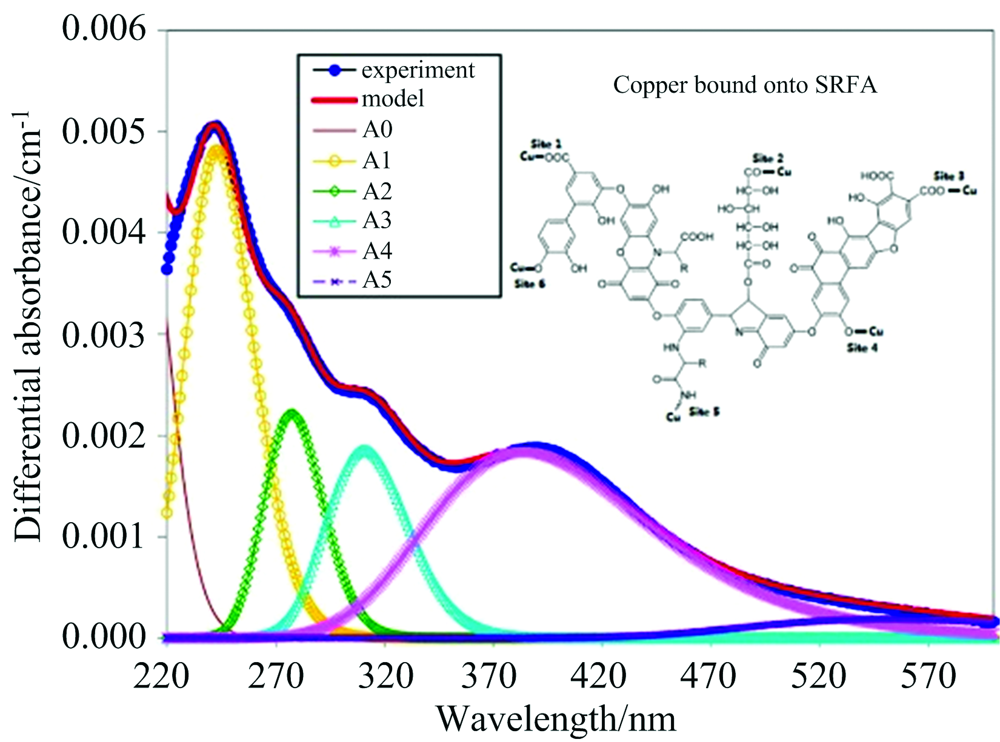
Yan等[25]通过紫外可见差分光谱法研究FA结构变化对FA与Cu2+络合过程影响, 并与模型化合物(磺基水杨酸、鞣酸和聚苯乙烯磺酸-马来酸等)的光谱进行差分吸收结果比较, 发现谱图中存在6个高斯谱带(A0, A1, A2, A3, A4和A5), 各谱带峰强最大值分别位于波长为208, 242, 276, 314, 378和551 nm处(图3); 模型化合物微光谱特征与实验真值FA特征不同, 但高斯谱带A1, A3及A2的特征在很大程度上归因于水杨基和多羟基酚基团的响应, 而高斯谱带A4和A5仅能在FA的差分光谱中检测到; 这表明Cu2+与FA中发色官能团的相互作用可能涉及不同官能团的去质子化(如水杨酸型基团和多酚羟基等); 该研究还发现Cu2+与FA中羧基和酚羟基键合的数量多于与凝胶静电相互作用键合; 同时, 在不同的Cu2+浓度体系下, FA的离散官能团、 整个电子跃迁系统、 FA分子构象会发生改变, 与铜络合引起的生色团相对位置也会发生改变; 不同FA分子之间的分子效应也对FA与Cu2+络合过程有一定的影响。
在FA与Cu2+的络合过程中, pH值通过改变FA活性位点对Cu2+的亲和力影响FA与Cu2+的络合, 主要由表面离子交换位点上Cu2+与H+之间的离子交换导致[26], 也可能是羧基在pH值介于2.5和7之间时解离形成羧酸根及酚羟基, 会在pH值为8和13.5之间解离形成(C6H5O-)[27], 随pH值的增加, FA表面的负电荷增加, 对Cu2+的静电吸引力增加, FA的络合能力也随之增加, 同时pH值也会影响游离Cu2+的浓度, 进而影响FA对Cu2+的络合量。
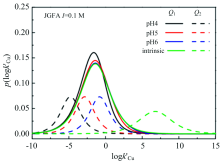
根据条件亲和谱(conditional affinity spectrum, CAS)可以描述络合剂在不同浓度下平衡状态亲和力或结合能的有效分布, Xu等[18]利用CAS对FA与Cu2+络合规律表征, 发现CAS值准确反映了pH值对羧基和酚羟基型络合位点对Cu2+亲和力的影响; 随着pH值的增加, CAS对羧基和酚羟基的亲和力都向较高条件迁移, 但是酚羟基向右迁移较明显, 与FA与Cu2+络合时质子交换所消耗的能量减少有关(图4)。
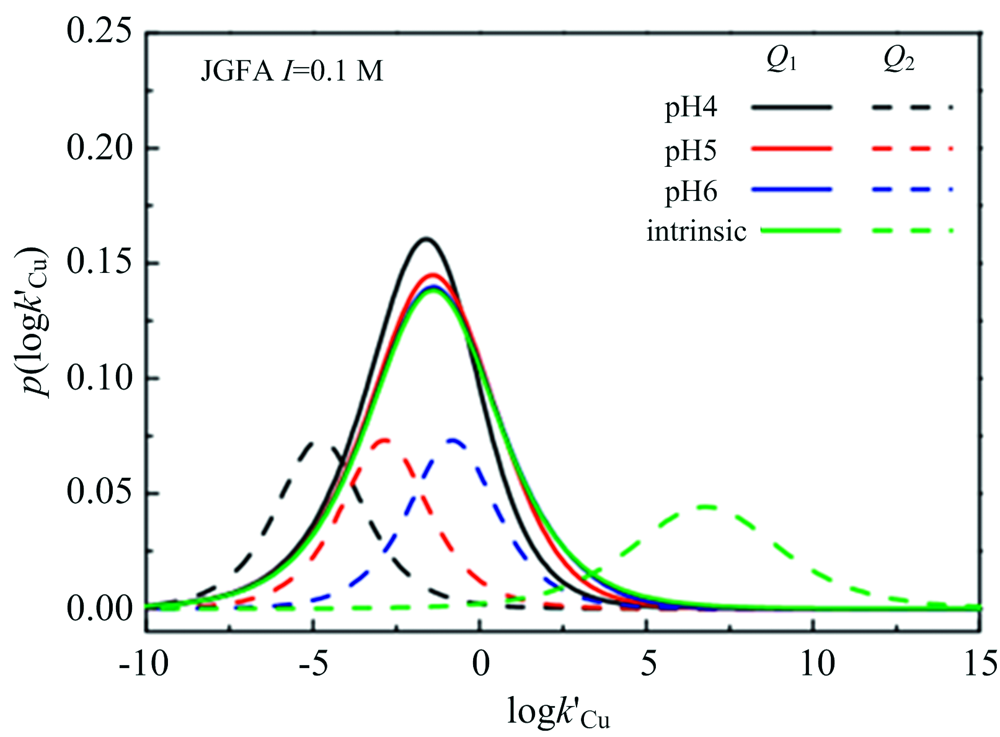 | 图4 不同pH值下CAS平均值的羧基和酚羟基分布的双对数拟合参数(logk′Cu表征不同铜离子浓度下活性位点对铜亲和力, p(logk′Cu)表征FA上活性位点分布)[18] Q1: 羧基; Q2: 酚羟基Fig.4 The logarithmic fitting parameters of the distribution of carboxyl and phenolic hydroxyl groups based on the average CAS value at different pH values (logk′Cu is a parameter related to the affinity of sites for copper under different copper ion concentrations, p(logk′Cu) is a parameter related to the distribution of the site on FA)[18] Q1: Carboxyl group; Q2: Phenol hydroxyl group |
实验揭示[28, 29]给定pH条件下, 与羧基位点质子相比, 需要较大的离子交换能量才能去除酚羟基位点质子, 导致pH值对酚羟基铜的影响比对羧酸铜的影响更大, 即随着pH值的增大, 酚羟基的作用逐渐减小, 羧基逐渐为控制FA与Cu2+络合的主要位点。 Zhang等[22]研究pH对FA与Cu2+络合过程规律时发现在pH值达到pHzpcp(电荷零点处的pH值)之前, 随着pH值从3升到4, FA的电离度也增大, FA表面产生更大的负电荷, 使得FA对溶液中阳离子的吸引力增大, FA的络合能力随着pH值的增大而增大, 此时Cu主要以带正电荷的Cu2+和[Cu(OH)]+形式存在; 当pH值高于pHzpcp时, 随着pH值从4增加到11, FA的络合能力显著下降, 这可能与Cu2+的水解和FA聚集体的形成相关(图5)。
由于来源、 生成途径以及后续的提取分级的多样性, 出现FA具有不同数量的酚羟基、 羧基以及不同的质子亲和力[22, 30, 31], 与Cu2+络合特征也不相同。
Joaquim等[32]用分子荧光光谱发现城市垃圾堆肥和畜禽粪便堆肥中提取的FA与从土壤中提取的天然FA结构组成上有一定的差异, 但与从低腐殖质土壤(如落叶层和上层土壤)中提取的FA有一定的相似性, 堆肥提取FA与天然土壤FA弱络合位点的光谱相似(< 420 nm), 而天然土壤FA强络合位点的谱带出现在较大波长(> 420 nm)处; 堆肥FA计算出的IcuL(结合位点荧光强度)都在30~40之间, 而天然FA则为0, 这表明在堆肥FA样品中, 约30%~40%的荧光基团不具有络合能力, 在天然FA中荧光基团几乎完全由络合结构构成, 而堆肥FA的络合位点的数量较少, 络合能力较低, 这可能与腐殖化程度较低有关[32, 33]。
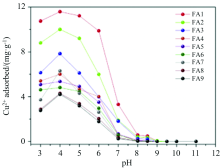
相较于从天然土壤提取的FA, 从堆肥污泥提取的FA相对强酸结构(pK值为3)浓度较低, 而这种相对强酸结构与水杨酸结构相对应, 在络合过程发挥积极作用, 使得两种FA在络合能力上存在一定的差异[33]。 在堆肥污泥提取FA中, 除含氧结构外, 含氮结合位点结构可能在络合反应中也起重要作用; 而天然土壤与Cu2+络合主要是由羧基和酚羟基结构引起的。 随着FA萃取次数的增加, FA有机碳、 芳香性、 分子极性、 羧基和酚羟基含量均降低, 分子量、 脂肪含量和含氮基团含量增加, FA与Cu2+的络合能力随之降低, Cu2+与FA不同萃取组分络合能力有明显差异, 但含氮基团对络合作用贡献随着萃取次数的增加而逐渐提高[22](图6)。
在FA与Cu2+的络合过程中, 溶液中存在的其他金属离子(Fe3+, Mg2+和Al3+等)与Cu2+发生竞争吸附, 同时, FA对Cu2+的络合能力随着溶液的离子强度(缓冲液中的离子如K+和Na+)的增加而下降, 研究认为, 可能与正电荷离子进入FA的电子层而增强电荷屏蔽效应有关, 使FA的静电吸附容量显著下降, 同时, 溶液中K+浓度增加会抑制Cu2+与FA的络合[34, 35, 36]。
Iglesias等[37]利用电位滴定测定FA与Cu2+和Ca2+的络合容量, 并对 Cu2+和Ca2+在FA上的竞争效应进行了深度分析。 研究指出, 随着Ca2+浓度的增加, 铜的络合程度降低, 而钙离子浓度越高, 铜的络合程度降低得越明显, 且Cu2+与FA络合比Ca2+与FA络合更依赖pH值, Cu2+所占据的位点数量仅占FA上电离酸基总浓度的20%。 Ca2+的竞争主要影响Cu2+-FA络合物的最大结合容量, 而对结合常数影响较小, 说明Ca2+与Cu2+在FA上的络合位点相类似, Ca2+会竞争FA上Cu2+的特定结合位点, 但对FA对Cu2+络合能力影响不大, 而Cu2+与FA络合更依赖于pH值是因为Cu2+比Ca2+在高pH值下更容易沉淀, 游离Cu2+比游离Ca2+浓度更少。
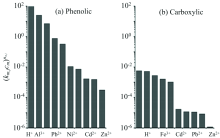
Rey-Castro等[29]利用CAS研究发现FA对不同离子亲和性各不相同, Al3+, Ca2+, Mg2+, Fe3+, Cu2+及Zn2+六种离子共存时, Al3+会优先与FA的酚类位点络合, Ca2+, Mg2+会对FA中羧酸类位点显示出更大的有效亲和力, Fe3+, Cu2+和Zn2+会与FA的酚类位点和羧酸类位点结合形成双齿类络合物[图7(a, b)]。 因此, 在存在其他金属离子的条件下, FA对Cu2+的络合能力的下降不仅仅归因于FA中Cu2+的特定络合位点被其他离子竞争性吸附, 也由于FA在其他离子的影响下对Cu2+静电吸引力的减小, 使得FA对Cu2+络合能力下降。
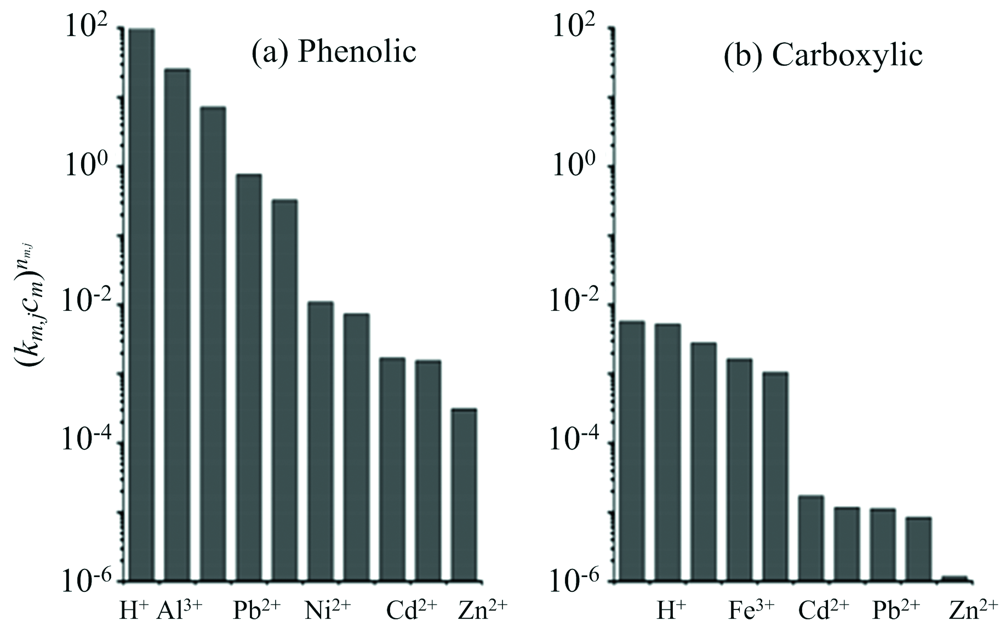 | 图7 (a)不同金属离子与FA酚羟基位点结合的(km, jcm |
Zhang等[38]通过逐步提取方法从湖泊沉积物中获得9种FA, 比较FA与Cu2+, Pb2+, Cd2+络合前后FTIR光谱, 3 447和1 630 cm-1附近的峰强度显著下降, 在1 120, 1 035和610 cm-1处峰强度下降, 表明FA的羧基、 酚羟基、 醇类、 多糖类物质、 以及含氮官能团参与了络合反应; 分别对610~620, 1 620~1 730和3 300~3 500 cm-1处FA与Cu2+, Pb2+和Cd2+络合前后的峰面积积分计算发现, FA与金属离子络合前后酚羟基的峰面积的差异为FA-Pb> FA-Cd> FA- Cu, 表明酚羟基对Pb2+的亲和性强于Cd2+和Cu2+; 而羧基和含氮基团峰面积表征的络合能力高低显示为Cu2+> Pb2+> Cd2+, 这表明这些基团对Cu2+的亲和力强于其他基团(图8)。
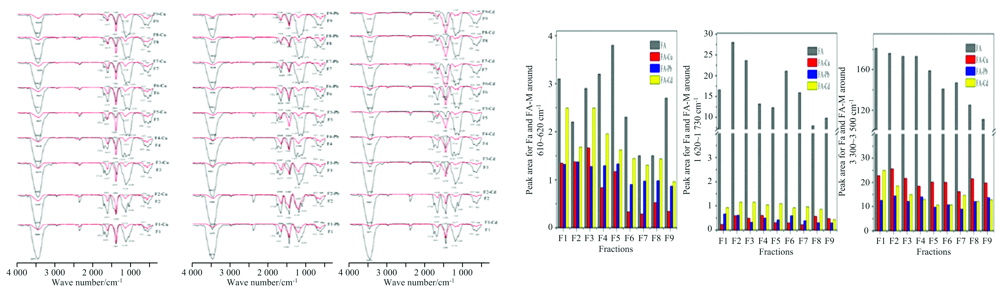 | 图8 依次提取的9种FA与Cu2+, Pb2+, Cd2+络合前后的FTIR谱[38]Fig.8 FTIR spectra before and after complexation of 9 kinds of FA with Cu2+, Pb2+, Cd2+[38] |
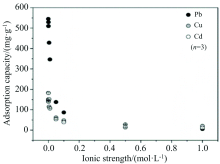
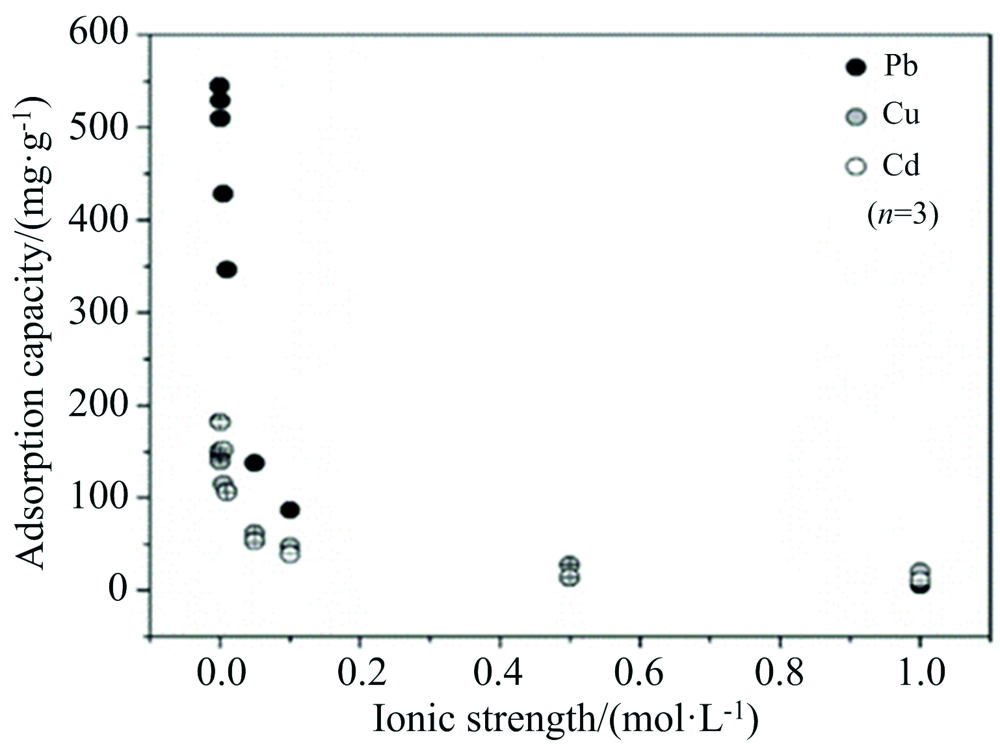
Li等[39]采用FTIR光谱对湖泊沉积物提取FA与重金属络合产物结构表征研究发现, 酚羟基、 羧基和含氮基团是提供重金属离子络合位点的主要官能团; 而FA对Cu2+, Pb2+, Cd2+的吸附量在较高离子浓度环境下趋于稳定, 相反在较低离子浓度(小于0.1 mol· L-1)时, FA吸附能力变化幅度大(图9)。 分析认为当离子强度增加时, 一方面阳离子会中和FA表面的负电荷, 使FA的双电子层被压缩, 降低了FA对金属离子的静电吸引力, 促使FA变成聚集体, 阻碍金属离子进入FA内部; 同时, 溶液中存在部分K+和Na+就近优先进入黄腐酸的电子层而增强电荷屏蔽效应, 与Cu2+, Pb2+和Cd2+发生竞争性吸附FA, 占据了FA表面酸性官能团(如羧基)的络合位点, 形成部分质子置换的钾盐, 黄腐酸对Cu2+的络合能力会随着溶液中离子如K+和Na+的增加而下降, 降低了FA吸附Cu2+, Pb2+和Cd2+能力[39, 40]。
红外光谱、 荧光光谱及差分光谱等现代检测技术, 对FA与Cu2+络合前后动态特征分析, 尤其是络合位点及络合过程规律揭示起到重要作用, 揭示了FA中的酚羟基、 羧基、 含氮基团、 pH值、 离子浓度以及FA的组分差异等对FA与Cu2+络合的重要影响, 有力促进了黄腐酸相关学科技术理论在现代农业、 生态修复及环境治理等多领域科学应用。 但是, 综合目前研究结果可见, FA与Cu2+络合机制研究中仍存在一些问题及挑战; 如, 目前对FA与Cu2+络合过程探索均是基于实验室层面数据进行分析, 对自然环境中FA与Cu2+络合应用研究非常欠缺; 相关作用过程规律仍不清晰和系统性; FA的分子结构对络合作用机理影响还不精准、 明确等, 因此, 面对黄腐酸相关在学科和行业领域迅猛发展的美好前景, 未来这些问题亟需实现有效突破, 以促进黄腐酸基础理论及应用的创新、 健康、 科学发展。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|