
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(NH4)2SO4, NH4NO3和(NH4)2SO4/NH4NO3混合气溶胶吸湿质量增长因子的快速测量方法
[李琼 , 马帅帅, 庞树峰, 张韫宏
, 马帅帅, 庞树峰, 张韫宏* ]
, 马帅帅, 庞树峰, 张韫宏]
|
|


作者简介: 李 琼, 1996年生, 北京理工大学化学与化工学院硕士生 e-mail: lq270224181@126.com
气溶胶颗粒的吸湿性决定了其尺寸、 浓度、 化学组成以及相态, 从而显著影响着全球气候、 大气异相化学以及人类健康。 运用在线、 原位、 连续扫描衰减全反射傅里叶变换红外光谱(ATR-FTIR)技术, 结合线性湿度(RH)控制系统, 实现了RH连续变化条件下气溶胶FTIR-ATR光谱的快速测量。 根据水弯曲振动谱带(~1 640 cm-1)峰面积随RH的变化, 得到了(NH4)2SO4, NH4NO3和(NH4)2SO4/NH4NO3混合气溶胶的质量增长因子(MGFs)、 潮解点(DRH)和风化点(ERH)。 与气溶胶的E-AIM模型预测值相比较, 实验结果表现出良好的一致性, 证实该方法是一种测量大气气溶胶MGFs, ERH和DRH的快速测量方法。
The hygroscopicity of aerosol particles determines their size, concentration, chemical compositions and phase states, and thus affects the global climate, heterogeneous atmospheric chemistry and human health. In this study, an on-line and in-situ rapid scan attenuated total reflection Fourier transform infrared (ATR-FTIR) technique coupled with a linear relative humidity (RH) controlling system was utilized to obtain the IR spectra of aerosols under different RH. The mass growth factors (MGFs), deliquescence relative humidity (DRH) and efflorescence relative humidity (ERH) of (NH4)2SO4, NH4NO3, and mixed (NH4)2SO4/NH4NO3 aerosols were determined rapidly by measuring the peak areas of the bending vibration band of liquid water (~1 640 cm-1). Comparisons between the measurements and the predictions from the E-AIM model showed good consistency, which verifies the rapid scan ATR-FTIR as a powerful tool for investigating hygroscopic behaviors and phase transitions of atmospheric aerosols. Furthermore, pure (NH4)2SO4 and NH4NO3 particles were found to effloresce at 49% and 25% RH, respectively, while mixed (NH4)2SO4/NH4NO3 aerosols with a mole ratio of 1∶1 and 1∶2 exhibited one-stage efflorescence transition beginning at 44% and 38% RH, respectively, upon dehydration. These results indicate that the presence of NH4NO3 can inhibit the crystallization of (NH4)2SO4, and formed (NH4)2SO4 seeds will act as heterogeneous nuclei to promote the efflorescence of NH4NO3 at higher RH. In addition, the double salt (NH4)2SO4·2NH4NO3 was formed upon efflorescence of mixed particles. These findings are critical for understanding complex phase transitions of mixed inorganic aerosols and interpretation for RH dependency of heterogeneous reaction rates of atmospheric reactive species.
水蒸气是大气的重要组成部分, 大气气溶胶是悬浮在大气中的微小颗粒物。 大气气溶胶与水蒸气之间存在着分配平衡过程, 当大气环境线性湿度(RH)升高或降低时, 气溶胶会吸水或失水, 以达到新的平衡, 这种特性被称为气溶胶的吸湿性[1]。 对于典型的无机盐, 当RH从高湿度区域开始降低时, 气溶胶液滴的组分浓度逐渐增大, 到达某个临界过饱和浓度时, 气溶胶由液态转化为固态, 此现象称为风化, 对应的RH为风化点(efflorescence relative humidity, ERH); 当RH由低湿度区域逐渐升高时, 无机盐固体颗粒物在低湿度阶段不会吸水, 当RH达到某一定值时, 固体颗粒物开始大量吸水变成液态, 此现象称为潮解, 对应的RH为潮解点(deliquescence relative humidity, DRH)[1, 2]。 DRH实际是无机盐饱和溶液对应的RH。 为了定量描述不同RH条件下特定无机盐气溶胶颗粒的含水量, 通常用质量增长因子(mass growth factors, MGFs)来描述气溶胶的吸湿性, MGF指某RH下颗粒物的质量与干颗粒质量的比值。 大气气溶胶吸湿性决定了颗粒物的含水量、 尺寸、 浓度、 相态、 化学组成和光学性质[3, 4, 5, 6, 7, 8, 9], 从而对辐射强迫、 大气能见度、 人体健康、 大气非均相反应、 PM2.5的非理想混合等产生显著影响[10, 11, 12, 13, 14, 15]。 研究大气气溶胶的吸湿行为是解决环境问题的重要步骤。
吸湿性研究有很多方法和实验手段, 如Liu等[16]建立了串联式微分迁移率分析仪(TDMA)来检测单分散气溶胶的吸湿性, 可以测量颗粒物的尺寸增长因子和DRH等。 Cziczo等[17]将亚微米级的气溶胶通入气溶胶流管, 依据红外光谱中无机盐离子的特征峰, 确定颗粒物的相态。 Ma等[18]用蒸汽吸附分析仪测量了一些微溶和不溶的无机盐颗粒物的吸水能力, 证明了蒸汽吸附仪可以精确测定颗粒物的吸湿增长, 甚至是颗粒物表面的水吸附层。 Chan等[19]报道了用电动态平衡(EDB)方法研究悬浮单液滴的吸湿性, 这种方法能悬浮几十微米尺寸的液滴, 可实时监测液滴的重量变化和拉曼散射信号, 从而能在分子水平上认识吸湿性与离子对结构的关系, 并能提供悬浮颗粒物相态的信息。
目前, 测量大气气溶胶颗粒物吸湿性的局限性, 主要体现在环境湿度动态变化条件下, 如何实时检测颗粒物的吸湿增长。 传统的吸湿性研究方法中, 一般需要先恒定RH, 待系统平衡后, 再测量此RH下的吸湿增长因子。 取决于RH的实验点分布密度, 吸湿性测试时间往往需要几小时甚至十几小时, 才能得到一组由高湿度到低湿度的吸湿增长因子实验数据。 快速测量吸湿性一直是人们关注的问题, Liang等[20, 21]利用高低湿度间的切换, 使EDB样品池内的RH连续变化, 同时利用硫酸液滴进行RH-时间曲线标定, 完成了对悬浮颗粒物吸湿性的快速测量。 该测量方法应用于(NH4)2SO4, NaNO3的质量增长因子测定, 结果与文献值吻合良好。 Ma等[22]将真空型傅里叶变换红外光谱仪与湿度脉冲变化系统联合, 能实现RH线性和脉冲式变化的控制, 在亚秒级分辨率采集光谱, 同时得到无机盐的含水量、 水蒸气等定量信息, 从而使吸湿性研究在几分钟内就可以完成。 但是真空傅里叶变换红外光谱仪价格昂贵, 装置的搭建需要有完整的压力方波系统控制湿度的变化, 整套实验装置造价高且操作复杂。 另外, 无机盐液滴中水的扩散系数约为10-5 cm2· s-1 [23], 气溶胶颗粒与环境中水蒸气的分配平衡时间约为毫秒级, 这为吸湿性快速测量方法奠定了基础。 若能实现湿度的线性变化和颗粒物含水量的同时测量, 就可以快速测定气溶胶颗粒物的吸湿性。 本文介绍了一种基于ATR-FTIR的气溶胶吸湿性快速测量方法。
本工作中, ATR-FTIR与湿度线性变化控制系统联合, 可以简便快速地完成对颗粒物吸湿性的测量。 同时, 红外光谱对颗粒物组分和相态的敏感性允许实时观测颗粒物的风化过程以及混合无机气溶胶中复盐的形成。 为了验证该方法, 对(NH4)2SO4, NH4NO3和(NH4)2SO4/NH4NO3混合气溶胶进行吸湿性测量, 在线性RH模式下确定了无机盐气溶胶的MGFs, ERH和DRH以及复盐的形成过程。
实验所用试剂(NH4)2SO4和NH4NO3均为分析纯(北京市通广精细化工公司); 所用溶剂均为超纯水(电阻率大于18 MΩ · cm); 载气为高纯N2(> 99.99%, 北京华通精科公司)。
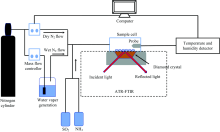
实验装置如图1所示, 红外光谱仪是德国Bruker公司的ALPHA-α 型ATR-FTIR。 自制的红外样品池放置在金刚石基底上方, 样品池RH由线性RH调节系统控制, 该系统由氮气瓶、 质量流量计、 高纯水罐和温湿度探测器组成。 质量流量计用来调节干湿氮气的流量, 总流量约为400 mL min-1, 进而控制样品池内气溶胶周围的环境RH, 另外通过对两个质量流量计进行编程控制, 可由软件调控样品池RH线性变化。 样品池内的RH值和温度由温湿度探测器实时测定, 探测器探头安装在样品池内部, 使RH和温度的响应时间大大缩短, 提高测量的准确性。 所有实验过程的RH范围约为0~80%, 温度约为23~26 ℃。
 | 图1 ATR-FTIR与湿度线性控制系统示意图Fig.1 Schematic diagram of an ATR-FTIR coupled with linear RH controlling system |
实验过程中, 首先通入干湿氮气约20 min, 将样品池内的空气排空, 然后测量红外光谱背景。 将样品溶液雾化, 雾化后的气溶胶液滴沉积在样品池的金刚石基底上。 将样品池封闭并通入约20 min湿氮气后, 调节两个质量流量计使样品池RH以0.5% RH· min-1进行线性变化, 同时连续测量不同RH下的气溶胶颗粒物的红外光谱。 每个改变红外光谱扫描8次, 扫描范围为4 000~500 cm-1, 分辨率为4 cm-1。
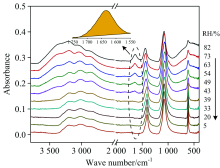
图2是(NH4)2SO4气溶胶在降湿过程中的红外光谱。 可以看出, 随着RH从82%降低到5%, 液态水的弯曲振动峰(~1 640 cm-1, 虚线区域)不断减弱, 峰面积(黄色填充区域)不断减小。 此峰面积可用来定量检测颗粒物的水含量, 进而确定气溶胶颗粒物在不同RH下的质量增长因子(MGFRH), 具体关系如式(1)
式(1)中,
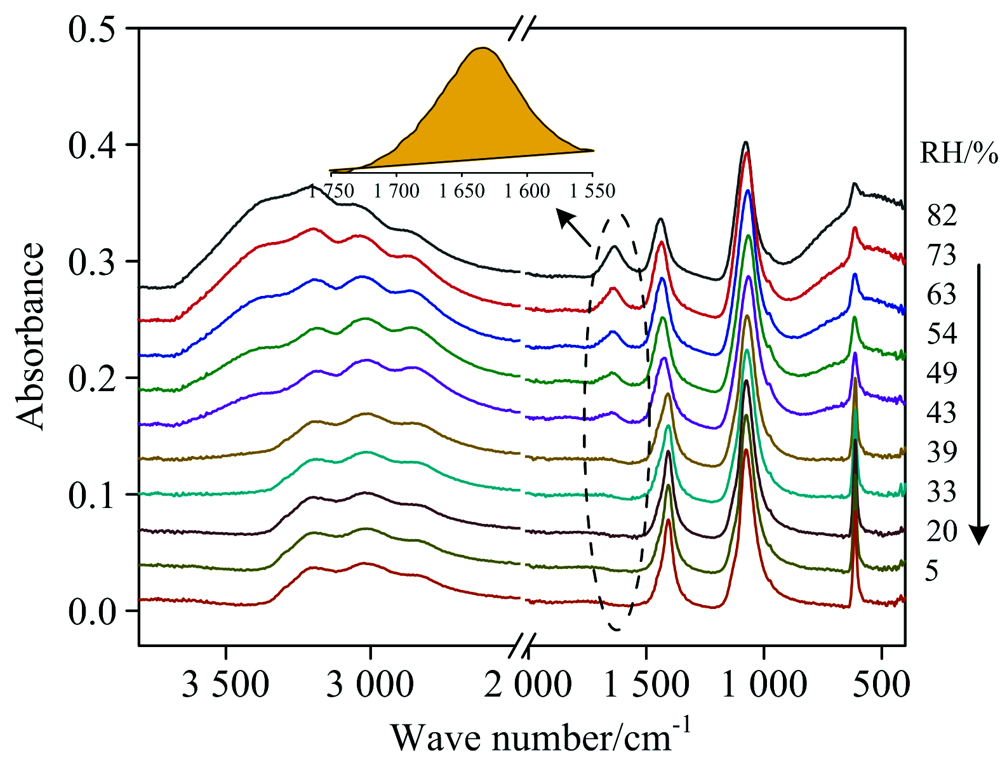 | 图2 降湿过程中(NH4)2SO4气溶胶的红外光谱Fig.2 IR spectra of (NH4)2SO4 aerosols during dehumidification |
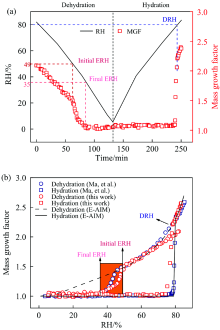
图3(a)是RH线性下降和上升过程中, (NH4)2SO4颗粒物MGFs随时间的变化关系。 首先, 在~81% RH时, 颗粒物的MGFs约为2.9, 此时颗粒处于不饱和溶液态。 随着RH下降, 水蒸气在气相和颗粒相之间的分配促使颗粒失水, 导致MGFs逐渐降低, 相对湿度低于80%以后, 颗粒物达到过饱和态。 当RH降低到49%时, 曲线出现一个转折点, 意味着(NH4)2SO4晶体开始形成, 49% RH被称为(NH4)2SO4气溶胶的初始风化点(initial ERH)。 随后颗粒物MGFs迅速下降, 在RH为35%时降至约1.0, 意味着(NH4)2SO4几乎完全结晶, 35% RH即是(NH4)2SO4的最终风化点(final ERH)。 因此实验测得的(NH4)2SO4的风化区间约为49%~35%。 在升湿过程中, 首先MGFs保持在~1.0, 说明颗粒物不吸水。 当RH逐渐升到79%时, MGFs急剧增加, 在~80% RH时增至~2.2, 表明(NH4)2SO4晶体完全潮解, (NH4)2SO4的DRH约为80%。 值得一提的是, 很多早期研究认为微米级的无机气溶胶在DRH之前也能溶解吸水, 当RH达到其DRH时, 无机盐晶体被完全潮解[24, 25, 26, 27]。 本工作将颗粒物完全潮解的RH值定义为颗粒物的DRH。
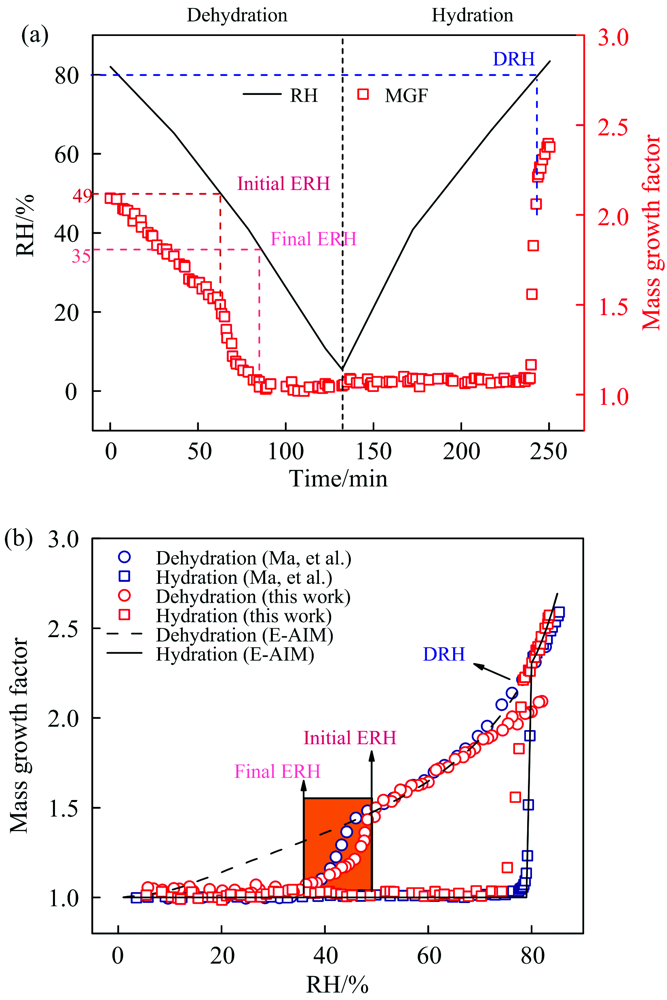 | 图3 (a) (NH4)2SO4颗粒的质量增长因子和环境RH随时间的变化; (b)(NH4)2SO4颗粒的吸湿曲线与文献值和E-AIM预测值的对比Fig.3 (a) Ambient RH and MGFs of (NH4)2SO4 as a function of time; (b) hygroscopic cycles of (NH4)2SO4 measured by this study and from literature and E-AIM predictions |
为了验证测量方法的准确性, 图3(b)将实验测得的MGFs与E-AIM模型预测值和文献值[22]进行比较, 结果表现出良好的一致性。 其中, 本工作测得的DRH与文献值和E-AIM模型吻合良好, 而初始ERH(~49%)高于(NH4)2SO4颗粒的均相成核ERH, 位于(NH4)2SO4的异相成核ERH范围(> 40%)[22]。 另外, 49% ERH略高于Ma等[22]测得的(NH4)2SO4初始ERH(~46%), 这主要是由于不同的颗粒沉积基底导致的, 即金刚石基底相比于氟化钙基底能诱导颗粒物在更高RH处发生风化。
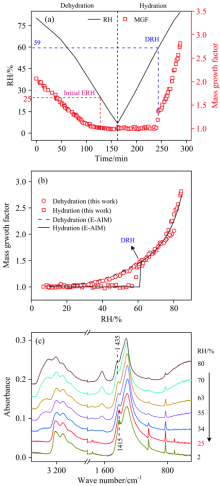
图4(a)是湿度循环过程中NH4NO3颗粒的MGFs随时间的变化。 随着RH逐渐降低, NH4NO3颗粒逐渐失水, 形成过饱和液滴, 但是MGFs在低湿度(< 25% RH)时也没有出现明显的转折点, 而是逐渐达到~1.0。 在早期研究中, Lightstone等[28]和Cziczo等[29]发现NH4NO3在降湿过程中没有出现结晶现象, 并且Cziczo等发现, NH4NO3在升湿过程中会从很低RH(< 10%)开始逐渐吸水, 表明NH4NO3颗粒可能与NaN
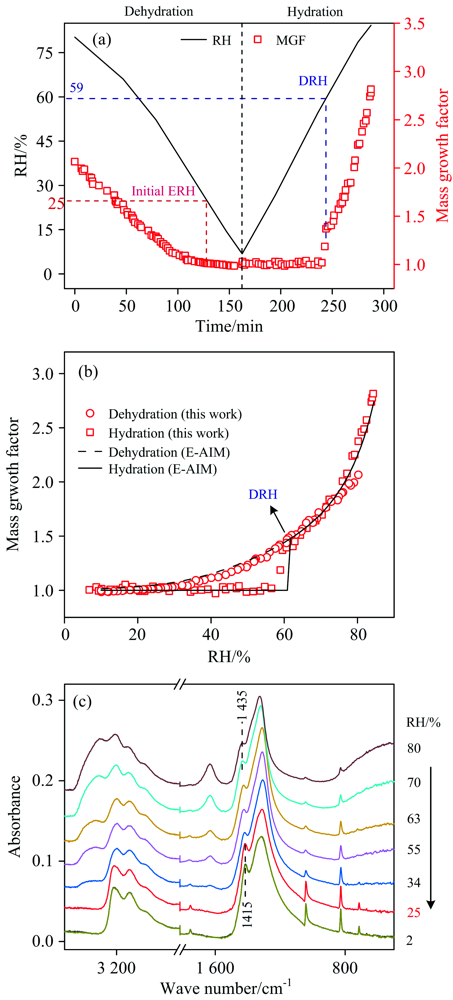 | 图4 (a)NH4NO3颗粒的质量增长因子和环境RH随时间的变化; (b)NH4NO3颗粒的吸湿曲线与E-AIM预测值的对比; (c)降湿过程中NH4NO3颗粒的红外光谱Fig.4 (a) Ambient RH and MGFs of NH4NO3particles as a function of time, (b) hygroscopic cycles of NH4NO3particles measured by this study and from E-AIM predictions, (c) IR spectra of NH4NO3particles during dehumidification |
为了进一步验证NH4NO3的风化过程, 将降湿过程中的NH4NO3光谱显示在图4(c)中。 在80% RH时, 1 435 cm-1的特征峰是溶液态
2.3.1 (NH4)2SO4/NH4NO3(物质量比1:2)混合气溶胶
图5(a)显示了RH线性变化时, 物质量比为1:2的(NH4)2SO4/NH4NO3混合气溶胶的吸湿过程。 随着RH线性下降, 混合颗粒的MGFs缓慢下降, 在38%~28%RH范围内从~1.5降至~1.0, 意味着混合颗粒发生了风化, 风化区间为38%~28%RH, 略低于Sun等[39]报道的硝酸铵摩尔分数为0.7(XAN=0.7)的(NH4)2SO4/NH4NO3混合颗粒的风化区间(40%~38% RH)。 图5(c)是降湿过程中(NH4)2SO4/NH4NO3混合颗粒的红外光谱变化。 在83% RH时, 1 440 cm-1的特征峰是溶液态
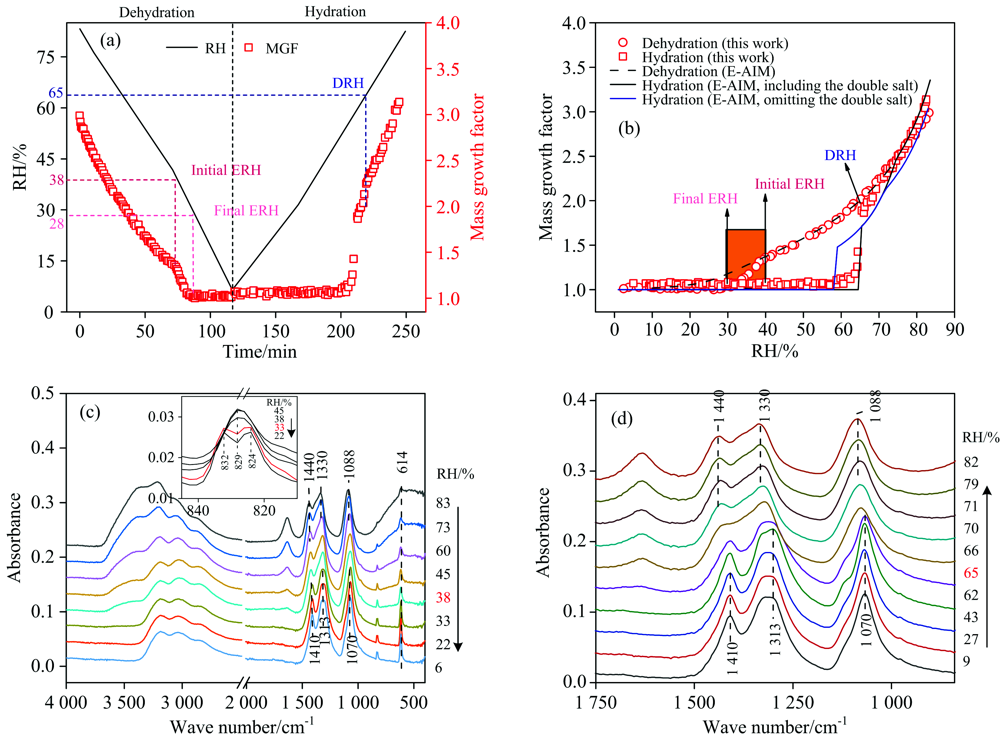 | 图5 (a) (NH4)2SO4/NH4NO3(摩尔比1:2)混合颗粒的质量增长因子和环境RH随时间的变化; (b) (NH4)2SO4/NH4NO3(摩尔比1:2)混合颗粒的吸湿曲线与E-AIM预测值的对比; (c) 降湿过程中(NH4)2SO4/NH4NO3(摩尔比1:2)混合颗粒的红外光谱; (d) 升湿过程中(NH4)2SO4/NH4NO3(摩尔比1:2)混合颗粒的红外光谱Fig.5 (a) Ambient RH and MGFs of (NH4)2SO4/NH4NO3 (1:2 mole ratio)mixed particles as a function of time, (b) hygroscopic cycles of (NH4)2SO4/NH4NO3 (1:2 mole ratio) mixed particles measured by this study and from E-AIM predictions, (c) IR spectra of (NH4)2SO4/NH4NO3 (1:2 mole ratio) mixed particles during dehumidification, (d) IR spectra of (NH4)2SO4/NH4NO3 (1:2 mole ratio) mixed particles during humidification |
在升湿过程中, 颗粒物MGFs首先保持不变[如图5(a)所示], 表明固态混合颗粒不吸水。 当RH达到65%时, MGFs开始急剧增加, 同时图5(d)中
2.3.2 (NH4)2SO4/NH4NO3(摩尔比1:1)混合气溶胶
图6是摩尔比1:1的(NH4)2SO4/NH4NO3混合颗粒的吸湿增长数据。 从图6(a)可以看出, 颗粒物MGFs随RH的线性下降而减小, 对应的ERH区间为44%~36%, 高于摩尔比为1:2的(NH4)2SO4/NH4NO3的ERH(38%~28%), 表明NH4NO3比例的增加会降低混合颗粒的ERH, 与Sun等[39]的研究结果一致。 另外, 纯组分NH4NO3的ERH约为25%, 而1:1 (NH4)2SO4/NH4NO3混合颗粒的初始ERH高达44%, 表明(NH4)2SO4的加入显著促进NH4NO3的风化结晶, 可能是由于(NH4)2SO4在44% RH时风化形成异相晶核, 进而诱导NH4NO3和(NH4)2SO4· 2NH4NO3在(NH4)2SO4晶种上发生异相成核。 随着RH上升, 颗粒物在~66%RH被完全潮解, 该DRH与Sun等[39]报道的XAN=0.6的混合颗粒的MDRH(~68%)接近, 并且本工作中1:1混合颗粒同样表现出了一阶潮解转变。
 | 图6 (a) (NH4)2SO4/NH4NO3(摩尔比1:1)混合颗粒的质量增长因子和环境RH随时间的变化; (b) (NH4)2SO4/NH4NO3(摩尔比1:1)混合颗粒的吸湿曲线与E-AIM预测值的对比Fig.6 (a) Ambient RH and MGFs of (NH4)2SO4/NH4NO3 (1:1 mole ratio) mixed particles as a function of time, (b) hygroscopic cycles of (NH4)2SO4/NH4NO3 (1:1 mole ratio) mixed particles measured by this study and from E-AIM predictions |
从图6(b)中可以看出, 实验测得的摩尔比为1:1的混合铵盐的MGFs与考虑复盐生成的E-AIM模拟结果相一致。 进一步证明了混合颗粒中复盐的生成, 以及ATR-FTIR搭载线性RH控制系统测量方法的准确性和实用性。
采用RH线性变化的吸湿性红外测量方法, 并对铵盐气溶胶的吸湿性进行研究, 测得的NH4NO3、 (NH4)2SO4及其混合气溶胶的MGFs, ERH, DRH与E-AIM模型值和文献值表现出良好的一致性, 证明了线性改变RH的FTIR方法对气溶胶吸湿性测量的高效性和准确性。
在中国北部的城市雾霾事件中, 硫酸铵和硝酸铵以及有机物占据了PM2.5颗粒的主要组分[45]。 其中硫酸盐和硝酸盐的吸湿增长, 往往决定了颗粒物的吸湿和相转变行为[46]。 同时, 硫酸盐和硝酸盐组分可以为大气异相反应提供重要的颗粒表面位点[47], 异相反应速率也与颗粒物环境RH密切相关[48]。 因此, 对于硫酸铵和硝酸铵混合颗粒吸湿性的研究, 有助于认识在重度污染事件中城市雾霾颗粒的组分、 相态和相转变过程, 并为大气异相反应对环境RH的依赖性提供一定的解释, 也为大气相关模型的理论模拟提供了有效数据。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|