
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
外电场对溴苯分子光谱特征影响的研究
[陈宇1  , 刘玉柱
, 刘玉柱1, 2, * , 王兴晨3 , 布玛丽亚·阿布力米提3, * ]
, 刘玉柱, 王兴晨]
|
|


作者简介: 陈 宇, 1996年生, 南京信息工程大学江苏省大气海洋光电探测重点实验室硕士研究生 e-mail: 20181217001@nuist.edu.cn
芳香族化合物是一类广泛存在于自然界的性质稳定、 具有很大毒性和致癌、 致畸性的化学物质。 其中, 溴苯(C6H5Br)是破坏自然环境并对人类身体健康有严重威胁的有机污染物之一, 研究外电场对溴苯光谱特征的影响在大气化学、 燃烧化学、 环境监测等领域有很广泛的应用。 基于密度泛函理论(density functional theory, DFT), 在BPV86/6-311G(d, p)水平上计算优化得到了不同外加电场(0~0.03 a.u.)下, 溴苯分子的红外光谱。 并在此基础上, 用同样的基组计算优化得到了溴苯分子的紫外-可见吸收(UV-Vis)光谱。 在实际测量过程存在诸多困难, 很难得到特定大小和方向电场下溴苯分子的红外光谱、 UV-Vis光谱等物理特性。 研究表明: 通过与实验值比较, 基于密度泛函理论计算得到的溴苯的红外光谱和UV-Vis光谱具有很高的准确度和很好的分辨率, 且包含的波长范围更广。 因此这些结果可作为实验值的补充, 为在理论上研究外电场对溴苯分子C—Br键振动频率、 红外光谱和UV-Vis光谱的影响提供了新的方法和思路, 具有很强的参考价值。 无外电场情况下, 由于C—H键振动, 溴苯分子的红外光谱在频率为718 cm-1处出现了最强吸收峰, 在1 455 cm-1处产生了一个仅次于最强峰的吸收峰。 C—Br键在727和1 185 cm-1附近有两个较强的吸收峰。 随着外电场从0增大到0.03a.u., C—Br键的两个红外光谱特征峰均发生不同程度的红移且振动强度增大, 部分C—H键振动也有类似的变化。 电荷沿C—Br键从6C原子转移到Br原子, 大大增强了Br原子的电负性, 6C原子的负电荷密度略有降低。 因此, 6C原子和Br原子之间的库仑力增大, 导致C—Br键红外光谱的振动强度增大。 同时, UV-Vis光谱最强峰对应的波长从191.6 nm蓝移到187.4 nm, 摩尔吸收系数从23 402.171增大到28 885.125。 这些结果为研究溴苯在外电场下的光谱检测提供了理论参考, 也对研究其他有机污染物的光谱检测方法有启示作用。
Aromatic compounds are widely existing in nature. They are stable, highly toxic, carcinogenic and teratogenic chemicals. Among them, bromobenzene (C6H5Br) is one of the organic pollutants that damage the natural environment and pose a serious threat to human health. The study of the influence of external electric field on the spectrum of bromobenzene has been widely used in atmospheric chemistry, combustion chemistry, environmental monitoring and other fields. In the present paper, density functional theory(DFT) at BPV86/6-311G(d, p) level are employed for the study of IR spectrum of bromobenzene in the external electric field. Based on the density functional theory, the UV-Vis spectra of bromobenzene molecules are obtained by the same method and basis set. In the actual measurement, it is difficult to obtain physical characteristics such as the infrared spectrum and UV-Vis spectrum of bromobenzene molecules under an electric field of a specified size and direction. The research shows that by comparing with the experimental values, the infrared spectrum and UV-Vis spectrum of bromobenzene calculated based on density functional theory have high accuracy and good resolution, and include a wider range of wavelengths. Therefore, these results can be used as a supplement to the experimental values, which provides a new method for theoretically studying the influence of the external electric field. Without the external electric field, due to the C—H bond vibration, the infrared spectrum of bromobenzene molecule has the strongest absorption peak at a wave number of 718 cm-1, and an absorption peak with an intensity second only to the strongest peak at 1 455 cm-1. The C—Br bond has two strong absorption peaks near 727 and 1 185 cm-1. As the external electric field increases from 0 to 0.03 a. u., both infrared absorption peaks of the C—Br bond are red-shifted and the vibration intensity increases, C—H bond vibrations have similar changes. The charge transfers the Br atom from the 6C atom along with the C—Br bond, which greatly enhances the electronegativity of the Br atom, and the negative charge density of the 6C atom decreases slightly. Therefore, the Coulomb force between the 6C atom and the Br atom increases, and the vibration intensity of the C—Br bond infrared spectrum increases. In addition, the wavelength corresponding to the strongest peak of the UV-Vis spectrum shifted from 191.6 to 187.4 nm, and the molar absorption coefficient increased from 23, 402.171 to 28, 885.125. These results provide a theoretical reference for studying the spectral detection of bromobenzene under an external electric field, and also have implications for studying the spectral detection methods of other organic pollutants.
分子中含有苯环的有机化合物叫做芳香化合物。 芳香族化合物是一类广泛分布于自然界的性质稳定、 具有很大毒性和致癌、 致畸性的化学物质, 在生物研究、 医药和农药生产、 化工合成以及环境保护等领域有很重要的影响[1, 2, 3, 4], 因此, 如何促进该类物质的检测和降解是近年来环境科研领域中非常具有实际和理论意义的课题之一。 在提高有机物降解效率的研究中, 国内外学者探索了各种不同的途径和方法[5, 6], 目前苯及其衍生物降解途径主要有光催化降解、 微生物降解、 水热降解等[7]。 然而外电场对溴苯光谱特征的影响还鲜有报道, 因此在这一方面的深入研究具有重要意义。
将溴苯分子置于不同强度的外电场(0~0.03 a.u., 1 a.u.=5.142× 1011 V· m-1)中, 首先使用DFT/BPV86方法在6-311G(d, p)基组水平上, 通过优化计算得到溴苯分子的基态几何结构, 计算了不同外加电场情况下溴苯分子的红外光谱。 然后在同样的基组6-311G(d, p)水平上采用杂化密度泛函方法TD-SCF-BPV86计算和分析了不同强度的外电场对分子UV-Vis光谱的影响, 为研究溴苯的光谱检测和电场降解方法提供理论依据。
当受到外电场作用时, 分子体系的哈密顿量表现为[8]
其中H0为无外电场时的哈密顿量, Hint为场与分子体系相互作用的哈密顿量, 在偶极近似条件下, 分子体系与外电场的相互作用能为[8]
其中, μ 为分子电偶极矩矢量, F为外电场矢量。
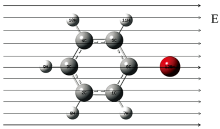
采用多种方法和基组计算优化溴苯分子结构, 发现BPV86/6-311G(d, p)方法优化的结果与实验值符合得最好, 优化得到的溴苯分子结构如图1所示[9, 10]。 所有计算全部在Gaussian09软件包中完成[11]。
 | 图1 溴苯分子稳定构型 箭头表示外加电场的方向Fig.1 Stable configuration of bromobenzene molecule The arrow indicates the direction of the applied electric field |
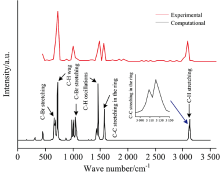
如图2所示, 在国际标准与技术学会化学电子数据库(NIST Chemistry Web-Book)中, 查找到溴苯分子在一定波长范围的红外光谱, 通过比较校正后的计算红外光谱与实验值, 发现两者具有相似的峰值频率和特征谱线, 其中在频率718 cm-1处出现了最强吸收峰(C— H键摇摆振动产生), C— Br键在频率为727和1 185 cm-1附近有两个吸收峰。 在1 455 cm-1附近, 由于C— H键振荡, 产生了一个仅次于最强峰的吸收峰。 苯环上C— C键的振动, 在1 570 cm-1处产生了一个较强的吸收峰。 在3 120 cm-1附近, 有4个C— H键振动叠加产生了多个强度不同的特征峰。 此外, 计算得到的红外光谱分辨率较高, 可作为实验值的补充, 为研究外电场下溴苯分子的红外光谱变化的提供参考。
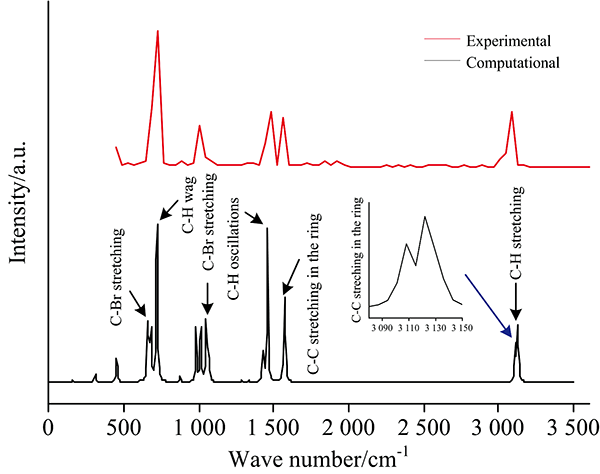 | 图2 实验测得的红外光谱(a)和校正计算后的红外光谱(b)对比Fig.2 Comparison of experimentally measured infrared spectra and corrected infrared spectra |
图3所示是不同外加电场对溴苯部分振动的红外光谱强度和振动频率的影响。 由图3(a)和(b)可以看出, 随着外电场从0增大到0.03 a.u., C— Br键的两个红外光谱特征峰均发生红移且强度增大, 其中一个吸收峰的峰值从43.4 a.u.增大到194.7 a.u., 对应频率从660 cm-1减小到593 cm-1; 另一个吸收峰的峰值从51.7 a.u.增大到363.6 a.u., 频率从1 015 cm-1减小到896 cm-1。 溴苯分子内电场的变化以及分子与外电场的相互作用两部分共同决定了溴苯在外电场作用下的总能量[12]。 随着外电场的增大, 电荷沿C— Br键从带正电6C原子转移到带负电的Br原子, 大大增强了Br原子的电负性, 6C原子的负电荷密度略有降低, 因此, 6C原子和Br原子之间的库仑力增大, 导致C— Br键拉伸红外光谱的伸缩振动强度增大。
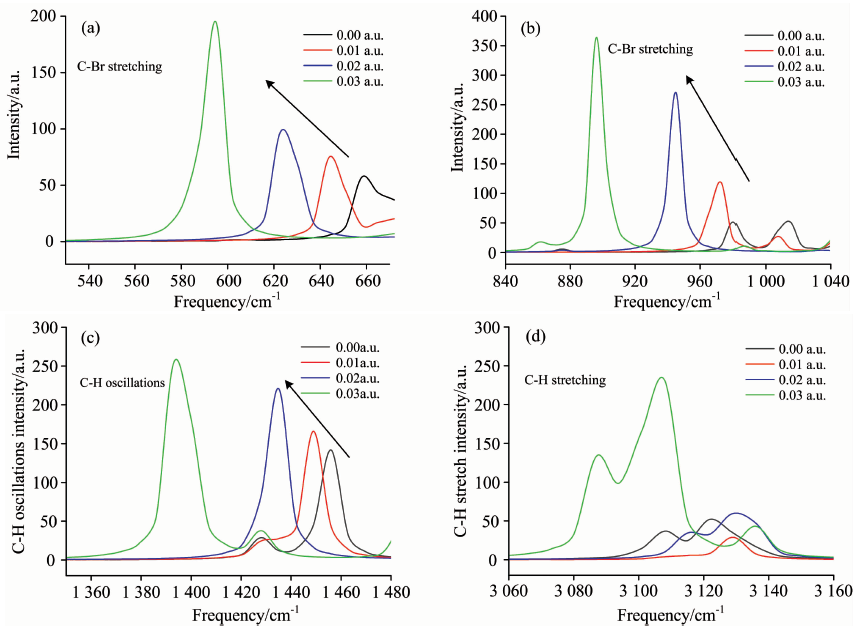 | 图3 外电场作用下的溴苯分子部分振动变化情况 (a): C— Br键伸缩振动; (b): C— Br键伸缩振动; (c): C— H键振荡; (d): C— H键伸缩振动Fig.3 Changes of some vibrations caused by external electric field (a): C— Br stretching; (b): C— Br stretching; (c): C— H oscillations; (d): C— H stretching |
图3(c)中, 由于C— H键振动, 在1 455 cm-1处产生了一个仅次于最强峰的吸收峰, 与C— Br键类似, 随着电场增大, 该吸收峰也发生蓝移且强度增大。 图3(d)中, 在3 120 cm-1附近, 5个C— H键振动产生了多个强度不同的特征峰, 这些峰排列密集, 并且随着电场变化, 变化规律较为复杂。
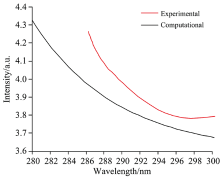
图4是当波长为在280~300 nm之间时, NIST Chemistry Web-Book中查找得到的UV-Vis光谱与计算得到的无外电场时溴苯分子的UV-Vis吸收光谱。 通过比较发现, 计算值与测量值非常接近, 因此, 通过计算绘制的波长在0~400 nm之间的UV-Vis光谱图(图5)具有很强的参考意义。
 | 图4 实验测得的UV-Vis光谱和校正计算得到的UV-Vis光谱对比Fig.4 Comparison of experimentally measured UV-Vis spectra with calculated UV-Vis spectra |
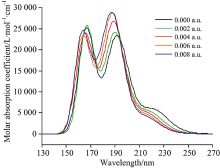
从图5中我们可以看出, 随着外电场从0增大到0.008 a.u., 其长波吸收峰峰值所对应的波长从191.6 nm蓝移到187.4 nm, 摩尔吸收系数从23 402.171增大到28 885.125。 短波吸收峰峰值对应的波长基本上没有变化, 但是吸收峰强度随着外电场增大而明显减小。
 | 图5 外电场作用下的UV-Vis光谱变化情况Fig.5 The UV-Vis spectrum changes under the action of an external electric field |
基于密度泛函理论, 研究了溴苯分子在外电场作用下红外光谱和UV-Vis光谱等物理性质的变化。 研究表明通过与实验值比较, 利用DFT方法获得的溴苯分子红外光谱和UV-Vis光谱具有较高的准确度和分辨率。 无外加电场时, 由于C— H键摇摆和拉伸振动, 在频率为718和1 455 cm-1处, 产生了两个强度较大的吸收峰。 C— Br键在频率为727和1 185 cm-1附近有两个吸收峰。 随着外加电场的增加, C— H键的两个主要的吸收峰红移且强度增大。 同时电场作用导致的电荷转移使得C— Br键的红外光谱振动强度逐渐增大, 对应波长出现红移。 UV-Vis光谱的最强吸收峰发生蓝移且强度增大, 另一个主要的吸收峰振动频率基本不变, 但强度减小。 这些结果为研究强电场下溴苯分子的光谱检测和研究提供了参考, 同时为其他有机污染物的检测方法和降解机理的研究提供了新的思路。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|