


{kind=link}
{kind=link}
{kind=link}
{kind=link}
不同温度下乙醇电氧化过程的原位红外光谱研究
[朱复春 , 涂昆芳, 李广, 姜艳霞
, 涂昆芳, 李广, 姜艳霞* ]
, 涂昆芳, 李广, 姜艳霞]
|
|


作者简介: 朱复春, 1988年生, 厦门大学化学化工学院博士研究生 e-mail: zhufuchun01@126.com
直接乙醇燃料电池因其优异的性能备受关注。 乙醇的电催化氧化并非简单的燃烧, 涉及多种催化反应过程。 乙醇的C—C键断裂选择性低, 以及乙醇氧化中间产物C1分子由于没有及时氧化离开催化剂表面而造成的催化剂中毒, 是制约其应用的瓶颈问题。 电化学原位红外光谱是在电化学反应的同时, 原位采集反应物种特定官能团的振动信息, 可在分子水平揭示反应过程, 推测反应机理。 不同温度条件下乙醇电氧化过程的研究, 有助于合理的设计高性能乙醇燃料电池催化剂。 选用高性能的PtRh/RGO催化剂, 结合同位素示踪法和电化学原位红外光谱技术, 研究了不同温度下乙醇的电氧化过程。 循环伏安研究表明, 乙醇电氧化性能及其C—C键断裂的程度为PtRh/RGO (45 ℃)>PtRh/RGO (25 ℃)>商业Pt/C。 电化学原位红外光谱从分子水平跟踪了乙醇的电氧化过程, 观察到随着电位的增加, CO2, CO, —CH3, —C—O特征峰的强度逐渐增加。 CO2和CH3COOH分别归属于乙醇完全氧化和不完全氧化的终产物, 因此红外光谱中两种物质特征峰积分面积的比值[CO2]/[CH3COOH]可做为CO2选择性的量度。 用来定量标定CH3COOH的特征峰是位于1 280 cm-1的—C—O振动峰, 但对于PtRh/RGO催化剂的红外光谱而言, 它的乙酸特征峰振动峰位1 280 cm-1附近出现1 214 cm-1甲醇衍生物的振动峰, 通过一种反射红外光谱与标样透射红外光谱差减扣除叠加峰方法, 定量计算了叠加峰中1 280 cm-1特征峰的积分强度, 从而计算出PtRh/RGO的CO2选择性。 结果表明对比25 ℃ 时, 45 ℃下PtRh/RGO具有更高的选择性, 0.3 V时提高48.1%, 0.5和0.6 V时略有提高, 0.4 V时降低, 这可能是乙醇中β-C和水中OH竞争吸附所致。 在两种反应温度条件下, CO2选择性都在电位高于0.4 V时呈现下降趋势。 为了进一步研究CO2来源于α-C或β-C的完全氧化, 使用同位素标记的13CH312CH2OH做为探针分子, 通过电化学原位红外光谱研究了25和45 ℃下PtRh/RGO电极上乙醇电氧化过程。 结果表明, β-C完全氧化为CO2的起始电位与温度无关, 都为0.3 V。 通过用13CO2/12CO2积分面积的比值定量分析, 发现45 ℃下, 该比值在电位0.3~0.5 V时相比于25 ℃下分别增加0.11, 0.18和0.22, 表明随着温度或电位的增加, β-C完全氧化的选择性增加。
Direct ethanol fuel cells are attracting much attention due to their excellent performance. Electro-oxidation of ethanol is not a combustion process, which involves multiple reaction processes. The low C—C bond cleavage ability and the poisoning caused by ethanol oxidation intermediate C1 molecules adsorb on the surface of catalysts are bottlenecks which restrict its application. Electrochemical in-situ fourier transform infrared spectroscopy ( in-situ FTIRS) is to collect the vibration information of the specific functional groups of the reaction species in situ and reveal the reaction process at the molecular level, helping to understand the reaction mechanism. High performance PtRh/RGO catalyst was used, to investigate the electro-oxidation of ethanol at different temperatures through the combination of isotope tracer and electrochemical in-situ FTIRS. Cyclic voltammetry studies revealed that the electro-oxidation properties of ethanol and the selectivity of C—C bond cleavage ability decreased in the order of: PtRh/RGO (45 ℃)>PtRh/RGO (25 ℃)>commercial Pt/C. Electrochemical in-situ FTIRS revealed the electro-oxidation process at the molecular level. It was found that CO2, CO, —CH3 and —CO characteristic bands increased gradually with the increase of potential. CO2 and CH3COOH are the products of complete oxidation and incomplete oxidation of ethanol, respectively. Therefore, the ratio of the integrated area of the characteristic bands in the infrared spectrum [CO2]/[CH3COOH] is the measurement of CO2 selectivity. The band at 1 280 cm-1 was used to quantitatively calibrate CH3COOH, but for the infrared spectra of PtRh/RGO catalyst, the superposition of band CH3COOH at 1 280 cm-1 and methanol derivative appeared at 1 214 cm-1. The superposition band subtraction method was developed to calculate the CO2 selectivity of PtRh/RGO. The selectivity of CO2 on PtRh/RGO at 45 ℃ was improved compared with that at 25 ℃, increased 48.1% at 0.3 V, slightly increased at 0.5 and 0.6 V, but decreased at 0.4 V, which might ascribe to the competitive adsorption of β-C in ethanol and —OH in water. At both reaction temperatures, CO2 selectivity show a downward trend at potentials above 0.4 V. To further investigate the complete oxidation of CO2 derived from α-C or β-C, isotopically-labeled13CH312CH2OH was used as the probe molecule, combined with electrochemical in-situ FTIRS to study the electro-oxidation of ethanol on PtRh/RGO electrodes at 25 and 45 ℃. The results show that the initial potential of complete oxidation of β-C is independent of temperature, both of which are 0.3 V. By quantitatively analyze the ratio of the integrated area of13CO2/12CO2, it was found that the ratio under 0.3~0.5 V at 45 ℃ increased 0.11, 0.18 and 0.22 compared with that at 25 ℃, which indicated that the selectivity of β-C increased with the increase of temperature or potential.
目前人类社会所面临的化石能源枯竭和严重的环境污染问题, 使得清洁、 无污染的绿色新能源成为国际研究热点之一[1]。 在可替代的环境友好能源中, 燃料电池是一种可直接且连续地把化学能转化为电能的发电装置[2]。 燃料经过催化反应, 而不是燃烧过程, 具有启动快、 比功率高、 结构简单、 工作温度低等众多优点。 对于燃料的选择而言, 乙醇不仅来源广泛、 低毒性、 能量密度高(8.1 kW· h· kg-1)、 易储存和运输, 而且可直接氧化, 较氢气燃料使用重整制氢装置, 乙醇燃料显著降低了成本[3]。 因此, 直接乙醇燃料电池(direct ethanol fuel cell, DEFC)倍受关注[4]。 然而乙醇通常电氧化生成不完全氧化产物乙醛和乙酸, 这使得乙醇电氧化的实际能量密度大大降低。 催化剂是能量转化的核心部件, 高选择性电催化剂可以有效提高燃料的利用率。
乙醇电催化氧化过程涉及多种反应途径, 产生多种不同的氧化中间物种或终产物[5]。 为了控制反应按照期望路径进行并且抑制副产物的生成, 深入认识复杂的电催化过程至关重要。 从分子水平研究电催化反应过程, 对于电催化剂的合理设计具有重要指导意义。 电化学原位傅里叶变换红外光谱(In-situ Fourier transform infrared spectroscopy, In-situ FTIRS)是在电化学反应的同时, 利用指纹图谱和表面选择规律, 实时获取电极表面吸附物种化学性质和涉及电化学试剂的溶液种类等信息的一种有效方法[6], 在揭示醇类燃料电池中的氧化反应过程, 推测反应机理方面具有独到之处[7, 8]。
使用具有立方体结构, 对于乙醇电氧化具有高选择性的PtRh/RGO催化剂, 用循环伏安法和原位傅立叶变换红外光谱法研究了不同温度下乙醇电催化氧化机制, 定量分析了不同氧化电位下CO2的选择性。 结合同位素标记示踪法与In-situ FTIRS技术, 进行了PtRh/RGO在不同温度下对于α -C和β -C的选择性的半定量研究, 评估了升高温度对于乙醇电催化氧化性能的提升。
采用以溴化钾作为形貌调控剂的多元醇法, 还原型氧化石墨烯作为载体, 合成了PtRh/RGO催化剂(详细过程参见文献[8])。
采用三电极电解池(包含工作电极, 对电极和参比电极), 连接于PAR-263A型恒电位仪进行电化学测试, 溶液为0.1 mol· L-1 HClO4+0.1 mol· L-1 C2H5OH。 电化学实验部分, 溶液的配制都使用超纯水, 溶液都预先经过氮气除氧预处理。
本工作所使用的是电化学原位红外反射光谱技术, 电解池为原位红外光谱薄层电解池。 该电解池的设计构造与三电极电解池类似, 不同之处在于底端为开口设计, 检测时用圆形氟化钙窗片对底端进行封口。 采谱过程中红外光源经镜面反射后穿透氟化钙窗片, 接收电极表面原位电化学测试过程中化学物质和吸附官能团的信息, 经电极表面反射并穿透氟化钙窗片后, 再经镜面反射到达红外光探测器进行光谱记录分析。
结果光谱为电位差谱, 即分别在研究电位ES和参考电位ER下采集单光束光谱(分别记录为R(ES)和R(ER)), 结果光谱根据公式Δ R/R=(R(ES)-R(ER))/R(ER)得到。 依据差谱公式定义可知, 向上的谱峰归属于参考电位下的物种, 而向下的谱峰归属于研究电位下的生成物种。
催化剂在45 ℃条件下的电化学原位红外光谱采用南京金慧策仪器科技有限公司的升温电极实现。 升温电极是通过对传统聚四氟乙烯包裹的玻碳电极进行加工, 通过内置微加热器和温度传感器, 实现加热和控温功能, 使得电极保持在不同温度下的电化学原位红外光谱的采集得以实现。
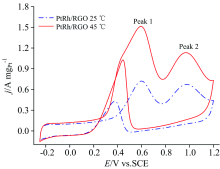
PtRh/RGO为还原氧化石墨烯负载的粒径约为10 nm, 表面具有缺陷位的立方体形貌合金纳米颗粒[8]。 电极通过预处理以后, 用电化学循环伏安法研究了PtRh/RGO催化剂分别在25和45 ℃条件下的乙醇电氧化性能, 如图1所示。 正向扫描时出现两个乙醇氧化峰, 峰1(Peak 1)归于乙醇不完全氧化生成的乙醛和乙酸, 以及完全氧化生成CO2; 而峰2(Peak 2)归于乙醇不完全氧化生成的乙醛和乙酸, Peak 1/Peak 2的比值可用于表征乙醇C— C键断裂的程度[9]。 在25 ℃时, PtRh/RGO的Peak 1电流密度达到0.67 A· mgPt-1而Peak 2则为0.62 A· mgPt-1, Peak 1/Peak 2为1.08, 优于商业碳载铂的0.82[10]。 这表明Rh的加入能显著提升Pt对于乙醇氧化的选择性。 在45 ℃时, Peak 1电流密度达到1.46 A· mgPt-1而Peak 2为1.07 A· mgPt-1, Peak 1/Peak 2为1.36, 比25 ℃条件下的比值提升了25.9%。 这表明, 随着温度的升高, PtRh/RGO催化剂对于乙醇C— C键断裂的选择性有所提升。
 | 图1 PtRh/RGO分别在25和45 ℃条件下乙醇电氧化的循环伏安曲线图(溶液为0.1 mol· L-1 HClO4+0.1 mol· L-1 C2H5OH, 扫描速率为50 mV· s-1)Fig.1 Cyclic voltammetric (CV) curves of PtRh/RGO for the electro-oxidation of ethanol at 25 and 45 ℃, respectively (0.1 mol· L-1 HClO4+0.1 mol· L-1 C2H5OH, scanning speed 50 mV· s-1) |
传统的电化学研究只能得到纳米电催化剂在电催化反应过程中整体的信息, 例如Peak 1和Peak 2都包含了多种氧化产物的叠加信息, 从而无法从分子水平深入理解电催化过程。 为了进一步论证升温使得催化剂对于乙醇完全氧化的选择性有所提升, 并研究乙醇在不同温度下的氧化过程, 我们引入升温电极, 结合电化学原位红外光谱技术, 详细研究了反应分子与纳米电催化剂表面相互作用的动态过程, 监测整个反应过程中的中间产物信息, 揭示电化学催化反应在不同反应温度下对不同产物的选择性。
图2为PtRh/RGO在45 ℃条件下, 参考电位为-0.25 V, 研究电位为0~0.6 V时的原位多步阶跃红外光谱图。 2 343 cm-1为乙醇完全氧化产物CO2的O=C=O振动峰, 一个乙醇分子分别含有一个α -C和一个β -C, 因此该二氧化碳可能同时包含两种C源完全氧化后的产物, 可以通过同位素示踪法对上述两种C源进行区分。 根据差谱公式, 研究电位下产生物种的谱峰方向向下, 由于CO吸附在纳米材料上表现出了异常红外效应[11], 使得CO的谱峰倒反, 在2 050 cm-1附近出现了向上的归属于研究电位下产生的线性吸附CO(COL)的振动峰和向下的参考电位下CO的振动峰。 可以看出Rh的加入并没有消除中间毒化物种CO(2 050 cm-1附近), 在电位为0 V时就已经有向上的研究电位下产生的CO2, 证明乙醇中C— C键的断裂已经发生, 并且随电位的增加, CO2产量逐渐增大。 这表明, 45 ℃ 时Rh的加入还可以在0 V电位下提供OH物种, 使得毒化物种CO被进一步氧化为CO2。 因此, Rh的加入不仅促进Pt对于乙醇C— C键断裂, 还可以使得毒化物种进一步完全氧化为CO2, 这与循环伏安法得出的结论一致。 1 720和1 370 cm-1的振动峰分别为乙醛或乙酸的— C=O和— CH3。 1 280 cm-1的振动峰归属于CH3COOH中的— C— O的振动峰, 因此可用1 280 cm-1特征峰(— C— O)定量计算CH3COOH。
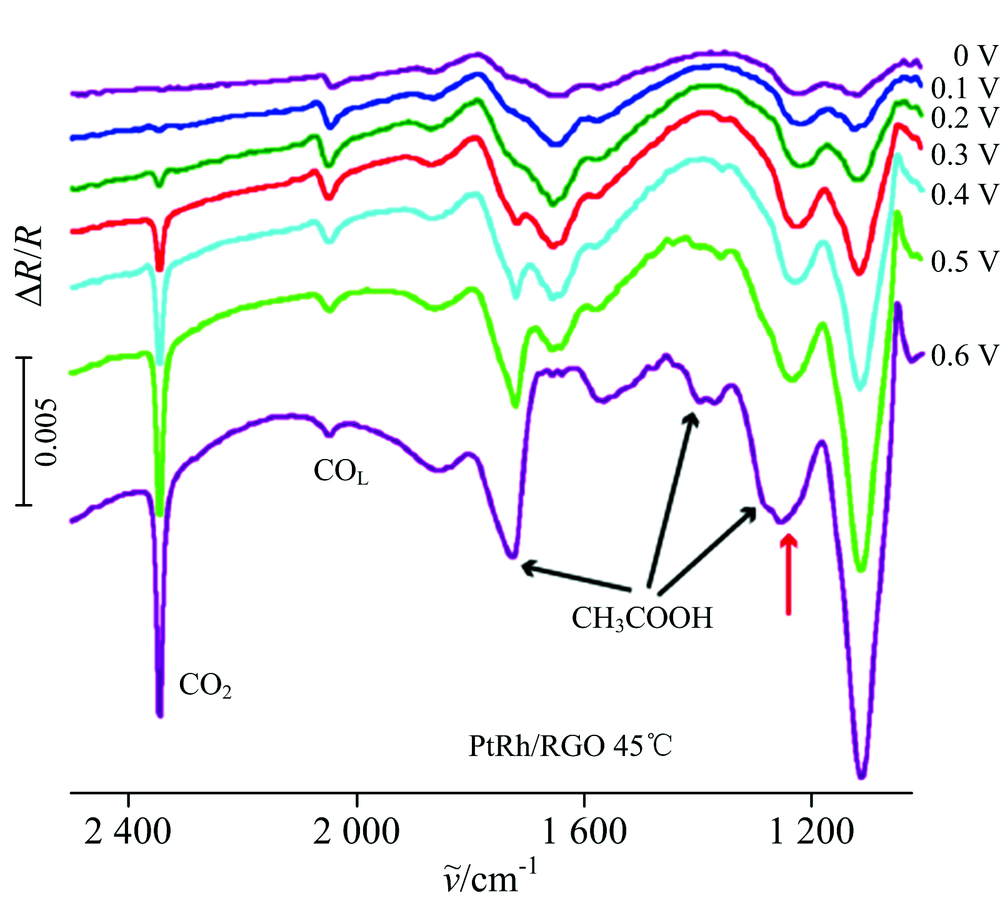 | 图2 温度为45 ℃时, PtRh/RGO在研究电位区间0~0.6 V内的原位多步阶跃红外光谱图, 参考电位为-0.25 VFig.2 In-situ MSFTIR spectra of PtRh/RGO at 45 ℃ in the potential range from 0 to 0.6 V, ER=-0.25 V |
通过对红外光谱的特征峰进行面积积分只能作为半定量分析指标, 想要直接对比催化剂在不同温度下对于乙醇完全氧化产生CO2的选择性, 通常用产物CO2和乙酸特征峰的积分面积比值(
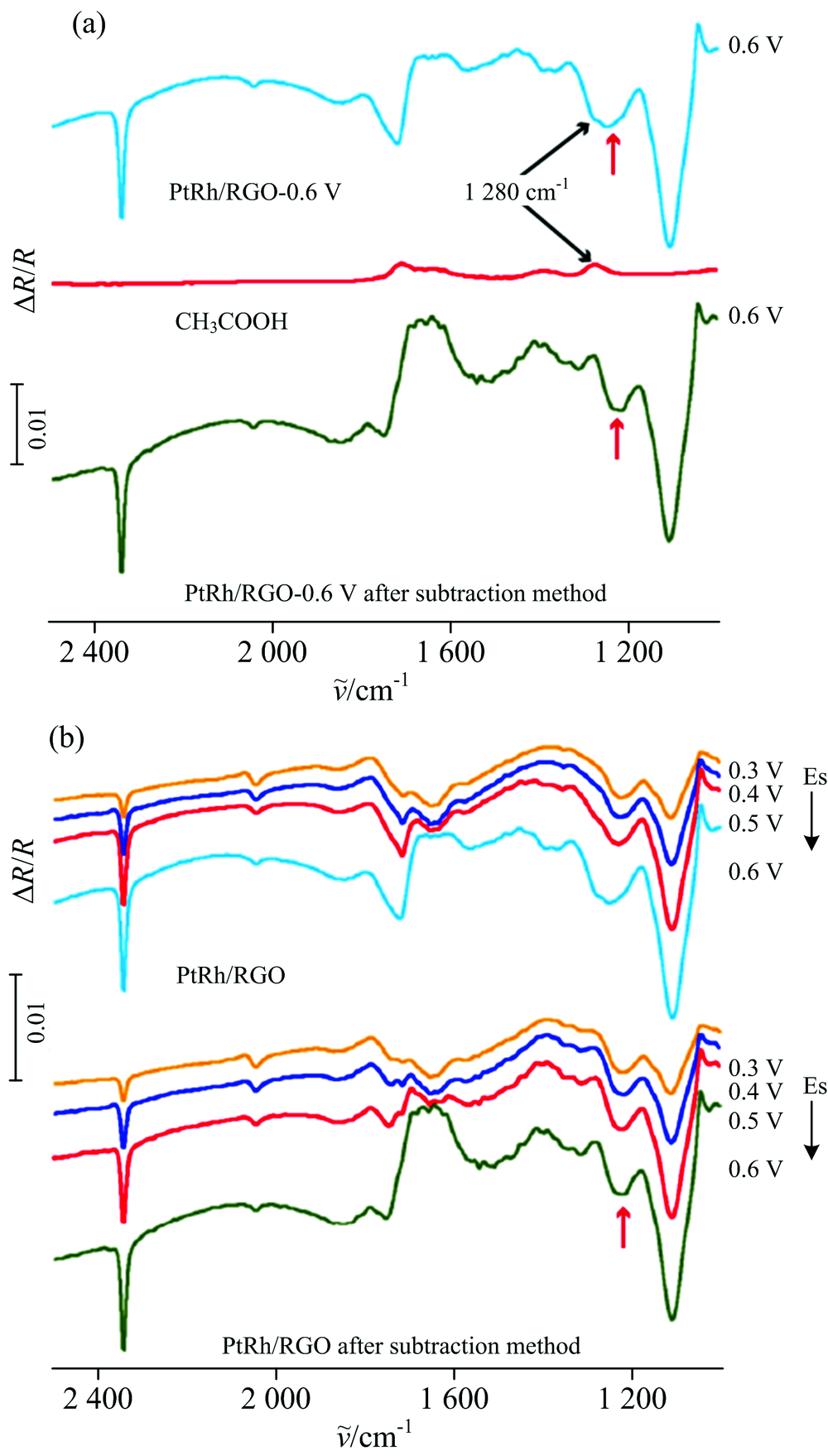 | 图3 (a)原位多步阶跃红外光谱图的叠加峰扣除法; (b)PtRh/RGO叠加峰扣除前后的原位多步阶跃红外光谱图(研究电位为0.3~0.6 V)Fig.3 (a) In-situ MSFTIR subtraction method; (b) In-situ MSFTIR spectra of PtRh/RGO before and after subtraction method (ES range from 0.3 to 0.6 V) |
以0.6 V电位下PtRh/RGO的原位多步阶跃红外光谱的叠加峰扣除法为例, 首先对20 mmol· L-1的乙酸标样溶液进行透射红外采谱, 然后将PtRh/RGO在0.6 V条件下的原位多步阶跃红外光谱与乙酸透射红外光谱进行差减, 使得位于1 280 cm-1的乙酸特征峰被差减掉, 仅留下位于1 214 cm-1的甲醇衍生物特征峰。 然后通过差谱前后, 叠加峰总积分面积与1 214 cm-1峰的积分面积进行差减, 即可得到被差减掉的位于1 280 cm-1的乙酸特征峰积分面积, 进而通过公式对CO2选择性进行定量分析。
图3(b)为PtRh/RGO在45 ℃条件下, 研究电位0.3~0.6 V范围叠加峰扣除前后的原位多步阶跃红外光谱图。 从图中可以明显看到, 叠加峰在扣除1 280 cm-1的乙酸特征峰以后, 仅剩下箭头所示1 214 cm-1的甲醇衍生物特征峰。 通过公式
| 表1 不同温度下PtRh/RGO的CO2选择性 Table 1 CO2 selectivity of PtRh/RGO under different temperatures |
对于乙醇电氧化研究而言, 如何提高产物CO2产率是研究重点。 前面介绍了, 乙醇分子中分别含有α -C和β -C两种可以被完全氧化为CO2的C源, 为了更进一步研究PtRh/RGO催化剂在不同温度下电催化乙醇产生CO2的过程, 使用C源标记的同位素示踪电化学原位红外光谱表征方法进行研究, 如图4(a)所示。 以同位素标记的乙醇(13CH312CH2OH)作为探针分子, 进行电化学原位红外光谱研究, 从图中可以看出, 无论测试温度为25 ℃或是45 ℃, 来自β — C完全氧化的终产物13CO2均在电位达到0.3 V时才开始产生。 这表明, 在上述研究温度条件下, 对于— CH3的完全氧化而言, 起始氧化电位与温度无关。
 | 图4 PtRh/RGO分别在25和45 ℃条件下, (a)电位区间0.2~0.5 V范围内的同位素示踪(13CH312CH2OH)原位多步阶跃红外光谱图, 参考电位为-0.25 V; (b)不同研究电位下的13CO2/12CO2比值Fig.4 (a) Isotopic tracer method (13CH312CH2OH) in-situ MSFTIR spectra of PtRh/RGO at 25 and 45 ℃ in the potential range from 0.2 to 0.5 V, ER=-0.25 V; (b) The ratio of 13CO2/12CO2 for PtRh/RGO at 25 and 45 ℃ under different potentials |
从图1中循环伏安曲线中可以看出, 随着温度的升高, CO2的总产量在相同电位下是显著增加的。 为了更进一步区分其中分别来自α -C和β -C完全氧化为12CO2和13CO2的占比, 分别对25和45 ℃反应条件下的13CO2/12CO2比值进行定量分析, 结果如图4(b)所示。 从图中可以看到, 当电位达到0.3 V以后, 都开始产生13CO2。 随着电位的增加, 13CO2/12CO2比值均逐渐增大。 这表明, 相同温度条件下, 随着电位的增加, β -C完全氧化为13CO2的占比逐渐增大。 另一方面, 对比相同电位下, 不同温度条件下的13CO2/12CO2比值, 0.3~0.5 V研究范围内该比值的增加量分别为0.11, 0.18和0.22。 可以发现, 在更高温度条件下该比值更大, 并且电位越高增加量也越大。 这表明随着温度的升高或电位的增加, β -C完全氧化为CO2的效率更高。
研究了高选择性PtRh/RGO催化剂在不同温度下的乙醇电催化性能, 结合电化学原位红外光谱表征技术和同位素示踪法, 对不同温度下的乙醇氧化过程进行深入分析。 发现PtRh/RGO催化剂在温度为45 ℃时, C— C键断裂产生CO2的能力较25 ℃时高。 为了对CO2选择性进行定量分析, 阐述了一种叠加峰扣除法, 用以区分1 280 cm-1的乙酸特征峰和1 214 cm-1的甲醇衍生物叠加峰, 分别进行峰面积积分计算, 从而计算出CO2选择性。 在0.3 V条件下, 提升温度时 CO2的选择性显著增加。 两种反应温度下, 当电位高于0.4 V时, 选择性都开始下降。 进一步通过同位素示踪电化学原位红外光谱表征, 发现25和45 ℃条件下PtRh/RGO乙醇电氧化过程中β -C完全氧化的起始电位与温度无关, 都为0.3 V。 随着温度升高或者电位增加, β -C完全氧化的选择性增加。 乙醇电氧化在燃料电池中做负极, 低电位下的阳极氧化使得燃料电池输出电压更高; 而燃料电池的实际工作温度通常高于85 ℃, 本研究表明低电位下升高温度有利于乙醇完全氧化, 并且乙醇中β -C完全氧化的占比增加。 该研究结果对燃料电池阳极催化剂的设计具有指导意义。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|