引用本文

赵省向, 宋雪燕, 邢晓玲, 李燕, 居学海. 高压下1,1’-二羟基-5,5’-联四唑二羟胺盐的振动光谱及同位素效应[J]. 光谱学与光谱分析, 2019,39(9): 2946-2952.
ZHAO Sheng-xiang, SONG Xue-yan, XING Xiao-ling, LI Yan, JU Xue-hai. Vibrational Spectra and Isotope Effect of Dihydroxylammonium 5,5’-Bis(Tetrazole)-1,1’-Diolate under High Pressure[J].
Spectroscopy and Spectral Analysis, 2019,39(9): 2946-2952.
Doi:10.3964/j.issn.1000-0593(2019)09-2946-07

Copyright©2019, 《光谱学与光谱分析》期刊社
《光谱学与光谱分析》期刊社 所有
1. 西安近代化学研究所, 陕西 西安 710065
2. 南京理工大学化工学院, 江苏 南京 210094
*通讯联系人
收稿日期: 2018-06-15
接受日期: 2018-12-12
Vibrational Spectra and Isotope Effect of Dihydroxylammonium 5,5’-Bis(Tetrazole)-1,1’-Diolate under High Pressure
1. Xi’an Modern Chemistry Research Institute, Xi’an 710065, China
2. School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
*Corresponding author e-mail:
xhju@njust.edu.cn
ZHAO Sheng-xiang, (1963—), research fellow, Xi’an Modern Chemistry Research Institute
Fund:National Natural Science Foundation of China (21207066)
IntroductionAn increasing attention has been paid to energetic salts in recent years since their overall properties could be optimized by the chemical modification of both ions[1, 2, 3, 4]. The coulomb interaction between anions and cations makes them have high density and stability compared with their atomically similar nonionic analogues. Niko Fischer et al. synthesized an energetic salts of dihydroxylammonium 5, 5’ -bistetrazole-1, 1’ -diolate (HATO). They found that it has a detonation velocity comparable to 2, 4, 6, 8, 10, 12-hexanitro-2, 4, 6, 8, 10, 12- hexaazatetracyclo[5.5.0.0.0]dodecane (CL-20) but lower sensitivity than commonly used high energy explosives such as cyclotetramethylene tetranitramine (HMX) and CL-20, indicating that HATO could be used as a promising insensitive high energetic material[5, 6, 7]. To probe insight into its microscopic factors of its low sensitivity, some investigations have been conducted to characterize the behaviors under high pressure and high temperature by theoretical as well as experimental methods. Dreger et al. used Raman spectroscopy to probe the stability of HATO and found that it transforms to two consecutive intermediates in heating[8]. An et al. studied the atomistic reaction mechanisms for the initial thermal decompositions of this system at high temperature by the quantum mechanics. They found that the protonated or deprotonated bistetrazole in the periodic system decompose under continuous heating, which eventually leads to the release of N2 and N2O[9]. The ionic energetic compounds are chemically stable over a broad range of pressure in comparison with their neutral analogues due to the strong Coulomb attraction between the counter ions, and some changes of the Coulomb attraction will take place at high pressure. It is reasonable to associate the shock initiation with the changes of interionic forces. The variations of vibrational spectra of HATO under high pressures as well as the deuteration effects were probed.
1 Computational detailsThe crystal structure obtained from the X-ray diffraction was used as the initial structure for the bulk (CCDC 872232). The unit cell of HATO was found to be a=0.544 1, b=1.175 1, c=0.656 1 nm and β =95.07° [5], crystallizing in space group P21/c (Fig.1). There were two irreducible anions and four cations in the unit cell. The generalized gradient approximation (GGA) method at the Perdew-Burke-Ernzerhof (PBE) and Perdew-Wang (PW91) levels and the ultrasoft psudopotentials were employed for the optimizations of cell parameters and molecular geometries, with dispersion force correction. Although the method includes the electron correlation, it is still computationally economic. The threshold for energy convergence is 2× 10-6 eV/atom in optimizing. The optimizations were performed with CASTEP code[10]. The optimized cell parameters are a=0.501 3, b=1.333 9, c=0.724 7 nm and β =100.00° at the GGA-PBE level, and a=0.543 5, b=1.224 6, c=0.682 5 nm and β =95.77° at the GGA-PW91 level. Therefore, the latter method is better and more suitable for HATO crystal. Consequently, all the geometrical optimizations for the crystal at high pressures were performed by the GGA-PW91 method. Since the deuteration does not change the electronic structure of the ions, the geometrical structures before and after the deuteration are identical. Then the vibrational frequencies were obtained by a single point calculation on the structures of ionic pairs from the optimized cell at high pressures. The non-periodic calculation of frequency was performed by the density functional theory method of Becke3-Lee-Yang-Parr (B3LYP) that produces consistent results with experiment[11, 12, 13]. The Gaussian 09 procedure was employed for the frequency calculations[14]. A scaling factor of 0.967 9 was adopted for the correction of frequencies[15].
2 Results and discussion2.1 Optimized crystal and molecular parametersThe optimized cell parameters at the GGA-PW91 level were listed in Table 1. The optimized cell parameters at both 0.0 and 0.1 GPa are consistent with the experimental values. On going from 0.0 to 40 GPa, the cell parameters of a, b and c decrease by 2.3%, 23.3% and 14.9%, respectively, indicating that the crystal is easily compressible along b and c axes compared to a axis. The pressure on a axis leads to two anions approaching with each other, thus a axis is less compressible. The crystal lattice energy is -1 015.71 kJ· mol-1 after corrected by dispersion forces. This large lattice energy is favorable for the stability of HATO and makes some contributions to its low sensitivity in response to mechanical stimulus. As can be seen from Table 2, the intermolecular distances change more than those of bond lengths. The intermolecular O…H distances generally decrease with increasing pressures, especially for the NO…HN distances. The C— C bond lengths slightly decrease with increasing pressures. However, the O— H and N— H bond lengths changes irregularly upon pressures. When the repulsion between these two atoms becomes too large upon pressure, the N— O internal rotation will occur to avoid an accumulated repulsion within O— H and N— H bonds. The variations of intermolecular distances and bond lengths bring about the shifts of vibrational frequencies as discussed below.
Table 1
Table 1
 Table 1 Optimized crystal structures of dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate under pressures| Pressure | a | b | c | β | V |
|---|
| 0.0 | 5.435 | 12.246 | 6.583 | 95.77 | 435.94 | | 0.1 | 5.443 | 12.181 | 6.585 | 95.98 | 434.20 | | 10.0 | 5.439 | 10.484 | 6.110 | 98.27 | 344.77 | | 20.0 | 5.379 | 9.862 | 5.916 | 99.48 | 309.51 | | 30.0 | 5.321 | 9.565 | 5.761 | 100.97 | 287.85 | | 40.0 | 5.307 | 9.388 | 5.600 | 103.35 | 271.47 | | Expt. | 5.441 | 11.751 | 6.561 | 95.07 | 417.86 |
Pressure in GPa; a, b and c in Angstrom; β in degree; V in Å 3. | Table 1 Optimized crystal structures of dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate under pressures |
Table 2
Table 2
Table 2 Some bond and hydrogen bond lengths of dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate under pressures| Pressure | NO…HO | NO…HN | C— C | O— H | N— H |
|---|
| 0.0 | 1.594 | 1.820 | 1.429 | 1.027 | 1.059 | | 0.1 | 1.599 | 1.810 | 1.428 | 1.026 | 1.060 | | 10.0 | 1.472 | 1.682 | 1.413 | 1.034 | 1.053 | | 20.0 | 1.445 | 1.630 | 1.400 | 1.032 | 1.040 | | 30.0 | 1.420 | 1.561 | 1.392 | 1.031 | 1.047 | | 40.0 | 1.437 | 1.542 | 1.384 | 1.027 | 1.029 |
Pressure in GPa; Atomic distance in Angstrom. | Table 2 Some bond and hydrogen bond lengths of dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate under pressures |
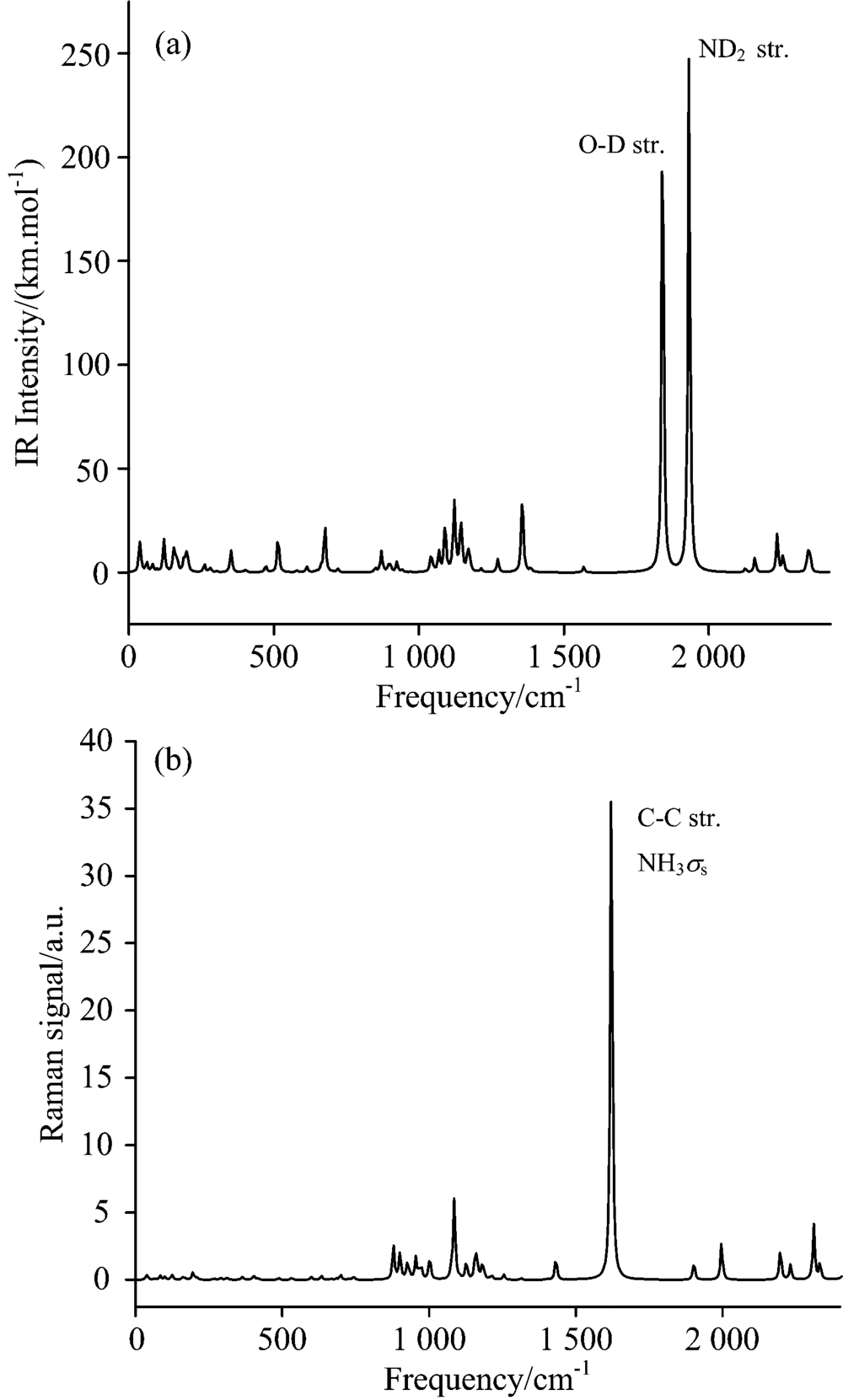
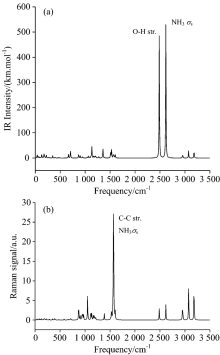
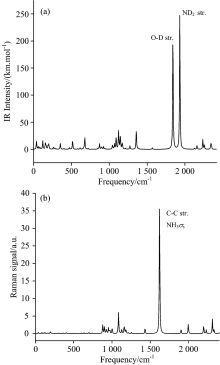
2.2 Vibrational assigns at ambient pressureThe overall IR and Raman spectra of HATO at standard pressure are shown in Fig.2 and those of its deuterated analogue are shown in Fig.3. Fifteen IR frequencies with large adsorption intensities before and after deuterium substituting at standard atmospheric pressure are listed in Table 3 and Table 4, respectively. The O1-H6 stretching of hydroxyl exhibits a very intense peak at 2 486 cm-1 which shifts to 1 841 cm-1 by deuterium substituting. The next intense peak at 2 616 cm-1 is assigned to symmetric deformation of protonated amino, which shifts to 1 931 cm-1 by deuterium substituting (deuteration). These large shifts of frequencies after deuterium substitution are mainly due to the great change of the reduced mass in deuteration. Classically, vibrational frequency for two atoms is inversely proportional to the root square of the corresponding reduced mass. Moreover, the shifts of frequencies lead to the changes of vibrational coupling.
Table 3
Table 3
Table 3 Vibrational modes, frequency and IR intensity of dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate| No. | Frequency | IR
Intensity | Vibrational Modes |
|---|
| ν 1 | 3 186.32 | 16.19 | H15-N14-H16 str. | | ν 2 | 3 178.68 | 13.98 | H3-N2-H4 str. | | ν 3 | 3 074.52 | 13.76 | H15-N14-H16 str. | | ν 4 | 3 071.47 | 16.09 | H3-N2-H4 str. | | ν 5 | 2 957.37 | 12.27 | O13-H18 str. | | ν 6 | 2 616.68 | 529.63 | H15-N14-H16-H17σ s | | ν 7 | 2 486.12 | 471.40 | O1-H6 str. | | ν 8 | 1 529.30 | 32.24 | H3-N2-H4 sci., O1-H6 r. | | ν 9 | 1 525.78 | 10.00 | H15-N14-H16-H17σ s, O13-H18 r. | | ν 10 | 1 509.70 | 23.20 | H3-N2-H4-H5σ s, O1-H6 r. | | ν 11 | 1 356.02 | 40.24 | C12-N8 str., C24-N20 str. | | ν 12 | 1 134.40 | 41.36 | ring breathing | | ν 13 | 900.99 | 10.07 | N10-N11 str., N22-N23 str. | | ν 14 | 867.98 | 10.95 | O1-H6 r. | | ν 15 | 705.38 | 26.75 | O13-H18 r. |
Frequency in cm-1. Abbreviation used: str, stretching; r, rocking; sci, scissoring; σ s, symmetric deformation; σ as, asymmetric deformation. | Table 3 Vibrational modes, frequency and IR intensity of dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate |
Table 4
Table 4
Table 4 Vibrational modes, frequency and IR intensity of deuterated dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate| No. | Frequency | IR
Intensity | Vibrational Modes |
|---|
| ν 1 | 2 348.89 | 8.18 | D15-N14-D16 str. | | ν 2 | 2 341.04 | 8.19 | D3-N2-D4 str. | | ν 3 | 2 235.80 | 16.53 | D15-N14-D16 str. | | ν 4 | 1 931.92 | 236.64 | D15-N14-D16 str., N14-D17 str. | | ν 5 | 1 841.09 | 223.76 | O1-D6 str. | | ν 6 | 1 357.05 | 37.16 | C12-N8 str., C24-N20 str. | | ν 7 | 1 169.16 | 9.32 | D4-N2-D5 sci. | | ν 8 | 1 145.81 | 11.47 | D15-N14-D17 r., D3-N2-D5 r., O13-D18 r. | | ν 9 | 1 123.00 | 19.86 | D3-N2-D4 sci. | | ν 10 | 1 120.57 | 12.65 | D15-N14-D16-D17σ as, ring breathing | | ν 11 | 1 090.83 | 22.93 | D4-N2-D5 sci., O1-D6 r. | | ν 12 | 1 069.23 | 8.60 | N10-N11 str., N22-N23 str. | | ν 13 | 1 042.54 | 7.91 | ring deformation | | ν 14 | 870.83 | 7.99 | N14-D15-D16-D17σ s | | ν 15 | 676.27 | 21.07 | ring breathing, O1-D6 r. |
Frequency in cm-1. Abbreviation used: str, stretching; r, rocking; sci, scissoring; σ s, symmetric deformation; σ as, asymmetric deformation | Table 4 Vibrational modes, frequency and IR intensity of deuterated dihydroxylammonium 5, 5’ -bis(tetrazole)-1, 1’ -diolate |
Frequencies in the range of 2 957 to 3 186 cm-1 belong to the O— H and N— H stretching of hydroxylammonium cation. There are weak peaks at 705 and 867 cm-1 for O— H rocking, which shift to 676 cm-1 by deuteration. As can be seen in Fig.2 and Fig.3, vibrational modes of H/D substitution at three highest frequencies are identical, while some low frequencies belong to different modes. Compared to HATO, one obvious difference in IR spectra after deuteration is that there exist weak peaks at 1 120~1 170 cm-1 for D-N-D scissoring/rocking or O-D rocking or ring breathing. The IR frequency of N-N stretching blueshifts from 900 to 1 069 cm-1 upon deuteration. Although these is no hydrogen atom in the anion moiety of HATO, the deuteration in cation still affects the vibrational mode of anion. The Raman spectra of HATO (Fig.2) and its deuterated analogues (Fig.3) look similar except for frequency decreasing upon deuteration.
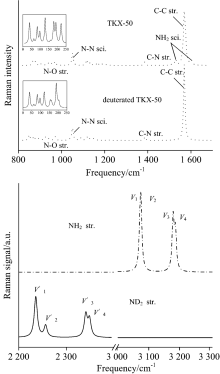
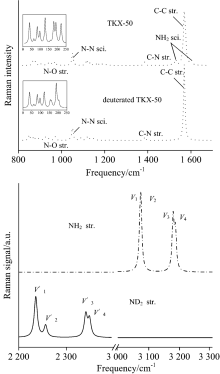
2.3 Isotopic effects on Raman spectraAs can be seen in Fig.4, the Raman spectra in the range of 800 to 1 600 cm-1 are generally similar as a whole with only one exception that there are two weak peaks on the opposite side of the strongest peak at 1 580 cm-1 for NH2 scissoring of HATO. However, these two peaks disappear upon deuteration. It is worthy to note that the strongest peak at 1 580 cm-1 is in good agreement with the experiment[8], indicating the computational method is suitable to the title compound. As can be seen in left amplified figures, a weak peak at 160 cm-1 increases its intensity, and a double peak at 180~190 cm-1 emerges into one peak with intensity decreasing upon deuteration. The vibrational modes of NH2 stretching ν 1/ν 2 at 3 180 cm-1 red shift upon deuteration, and ν 3/ν 4 at 3 090 cm-1 of HATO also red shift and at the same time split into two peaks upon deuteration. The calculated isotopic ratio, ν (NH2)/ν (ND2) for ν 1 to ν 3 modes are in the range of 1.36~1.38, which are consistent with the value of 1.39 obtained from the ratios of reduced masses for these atoms. However, ν (NH2)/ν (ND2) for ν 4 mode is as large as 1.59 since the ν 4(NH2) and ν 4(ND2) modes are not identical with modes coupling involved in the latter.
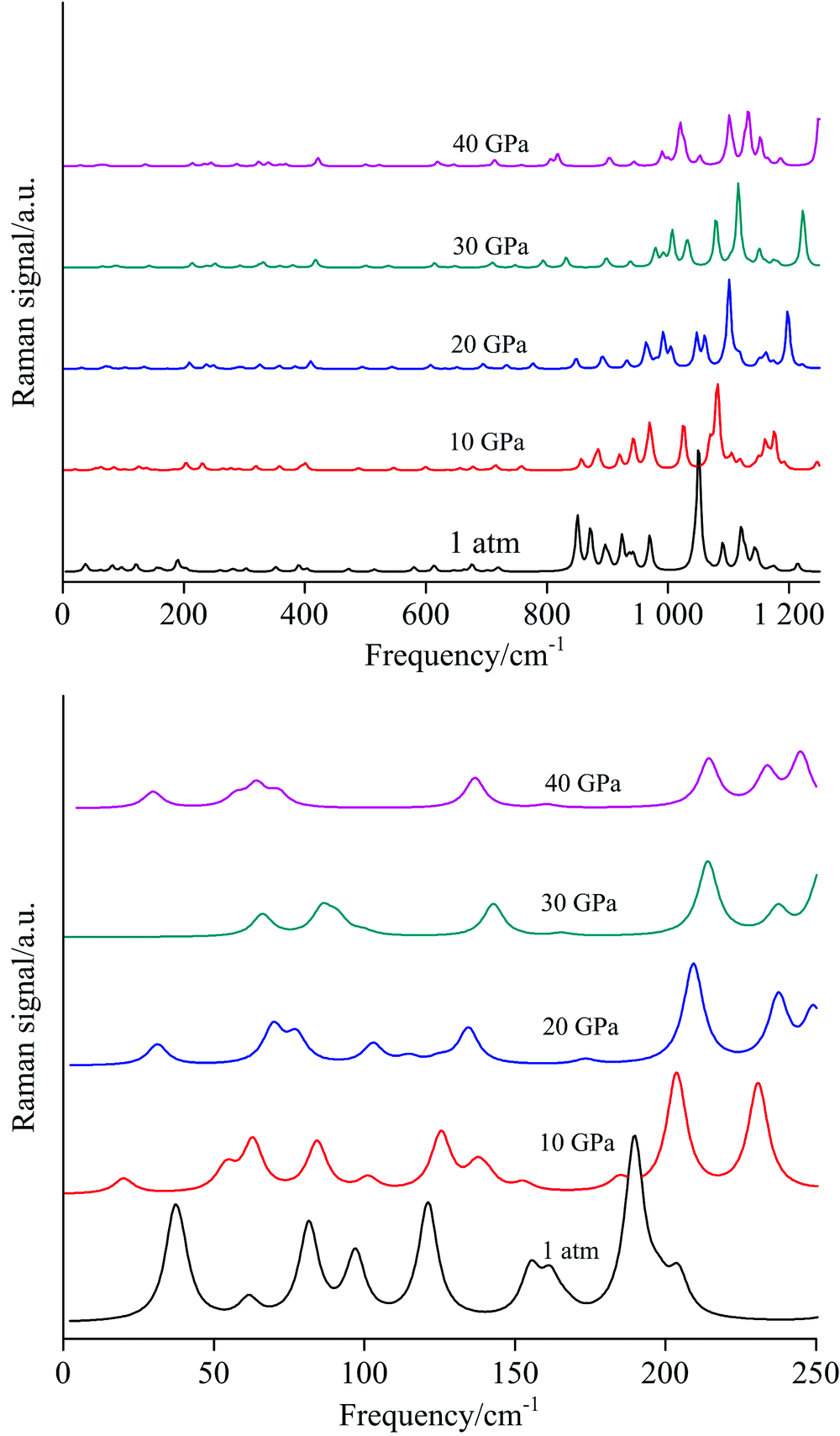
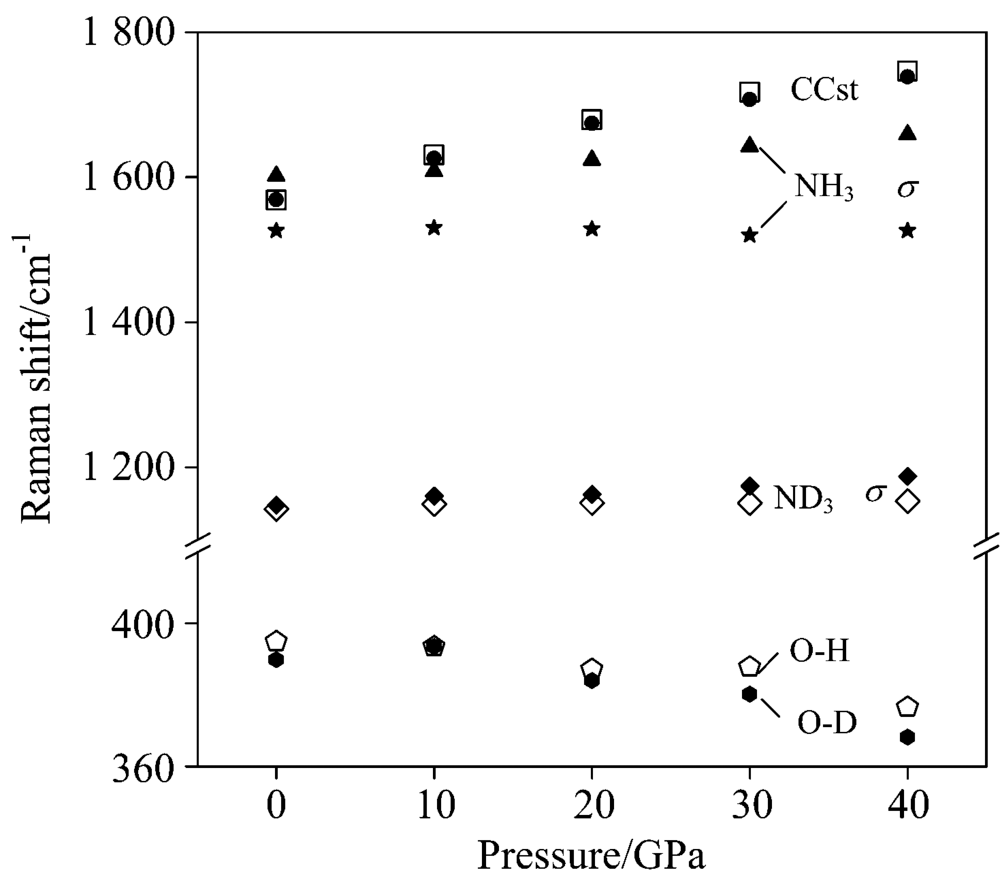
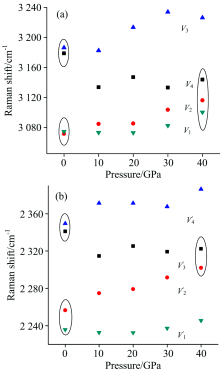
2.4 Spectra at high pressuresFig.5 displays the Raman spectra in pressure. In general, almost all the peaks blueshift as the pressure increase. The atomic equilibrium distances decrease when high pressure is imposed. This leads to the strengthening of chemical bonds as compared to those under standard pressure. However, the blueshift becomes smaller and smaller when the pressure comes up to 40 GPa since the nuclear repulsion between neighbor atoms increases dramatically as their distance decreases, i.e., the compressibility of chemical bonds become smaller. The Raman shifts vs. pressure is shown in Fig.6. The Raman shifts for C— C stretching increase as the pressure increases. For two NH3, σ deformations, one shift increases and another hardly change with increasing pressure. However, both the ND3, σ deformations hardly change with increasing pressure. For O— H and O— D vibration modes, the Raman shifts decrease as the pressure increases. The intermolecular hydrogen bond O19-H6 increases as the pressure increases. Consequently, the O1-H6 bond weakens with increasing pressure. Fig.7 shows the Raman shifts of the NH2 and ND2 stretching modes ν 1 to ν 4 under pressure. The Raman shifts of the ν 1 and ν 2 modes for NH2 increase slightly with pressure increasing up to 20 GPa, but increase greatly over 30 GPa. The Raman shift of ν 3 mode increases but that of ν 4 mode decreases as a whole, leading to the ν 1/ν 2/ν 4 modes coupling at high pressure as indicated by the oval circle in Fig.7. Upon deuteration, the most characterized change of Raman shifts for ND2 is that the ν 2 stretching mode increases dramatically with high pressure as compared to those of NH2, which leads to a ND2ν 2/ν 3 modes coupling at high pressure.
3 ConclusionsThe periodic density functional theory calculation together with ultrasoft psudopotentials reproduced the experimental crystal structure of HATO and was thus employed for the optimizations of both molecular structure and cell parameters. Based on the optimized crystal structure of HATO under high pressures up to 40 GPa and the non-periodic calculations of frequencies with a scaling factor of 0.967 9, the IR and Raman spectra at different pressures were predicted. The intermolecular O…H distances generally decrease with increasing pressures. However, the O— H and N— H bond lengths changes irregularly upon pressures. The predicted strongest Raman peak at 1 580 cm-1 involving C— C stretching and NH2 symmetric deformation is in good agreement with the experiment, indicating the method is suitable for the title system. Although there is no hydrogen atom in the anion moiety of HATO, the deuteration in cation still affects the vibrational mode of anion. For O— H and O— D vibration modes, the Raman shifts decrease due to the strengthening intermolecular hydrogen bond as the pressure increases. The calculated isotopic ratio, ν (NH2)/ν (ND2) for ν 1 to ν 3 modes are in the range of 1.36~1.38, which are in consistent with the value from the ratios of reduced masses for these atoms. The couplings of vibrational modes change with both deuteration and pressure. The results provided valuable information for understanding the properties of high energetic materials under high pressure.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 ]
]