{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函的茶多酚分子EGCG和GCG的光谱计算
[于建成1  , 唐延林
, 唐延林1, * , 常瑞2 , 魏晓楠1 , 袁荔1 , 袁园1 ]
, 唐延林, 常瑞|
|


作者简介: 于建成, 1990年生, 贵州大学物理学院硕士研究生 e-mail: 18085112281@163.com
茶多酚是绿茶中主要生化活性成分之一。 选取茶多酚中含量较高, 同时也是性质较活泼、 功效较明显的表没食子儿茶素没食子酸酯(EGCG)及其异构体没食子儿茶素没食子酸酯(GCG)分子做红外光谱和紫外光谱的计算和研究。 使用Gaussian软件, 采用B3LYP密度泛函理论(DFT)在6-311g(d,p)基组水平上优化其几何构型。 频率计算得到红外光谱后, 再进行振动特征分析, 可以看到在EGCG和GCG的红外光谱图中每个振动模式下所有基团振动的权重, 结合谱图做出相应的振动归属和对比分析。 发现: 两分子红外谱图相似, 分别在1 711和1 717 cm-1处为羰基的伸缩振动吸收峰, 苯环上酚羟基的伸缩振动吸收峰集中在3 500~3 800 cm-1, 1 000~1 600 cm-1的多个峰都有苯环面内弯曲振动参与, 在1 350和1 280 cm-1附近吸收峰是亚甲基次甲基面内弯曲振动引起的, 在500 cm-1以下吸收峰都为原子的面外弯曲振动。 采用固相粉末压片法, 使用IRPRESTIGE-21红外光谱仪测量了EGCG分子的红外光谱(400~4 000 cm-1), 对比理论计算的EGCG分子红外光谱各吸收峰位值, 发现在固相中实际测得的EGCG分子的红外光谱与气相下的理论计算值基本吻合, 理论计算值略微有些红移, 原因可能是理论计算在气相条件下采用的势函数存在误差, 相比于无分子相互作用力的气相, 实际测量固相光谱的分子键强度比气相条件下要略大些。 使用Gaussian软件, 采用含时密度泛函理论(TD-DFT), 选取乙醇作为溶剂, 计算了EGCG分子的15个激发态, 分析了激发态的组成和能级跃迁情况。 计算所得的2个吸收峰分别位于229.3和276.4 nm处, 主要对应p电子与苯环π键上电子形成的p-π共轭的电子跃迁及苯环、 杂环上π→π*跃迁。 从分析振子强度得知, 基态跃迁到S4, S5, S6和S12激发态为产生紫外光谱的主要原因, 另外的激发态可能为禁阻跃迁, 振子强度均小于0.01。 上述计算值与使用UV-6100S型紫外分光光度计所测得的EGCG分子在乙醇溶剂中235.1和278.7 nm的最大吸收峰吻合, 计算值略有蓝移, 可能是茶多酚提取时或本身就带有弱碱性所致。 该研究可为研究EGCG分子和GCG分子的性质和生物活性及茶多酚的抗氧化性提供理论参考。
Tea polyphenol is one of the main biochemical active components in green tea. Being a higher content of components and more active and effective ingredient in tea polyphenols, the epigallocatechin-3-gallate (EGCG) molecule and its stereoisomer GCG are selected to calculate and study the infrared spectrum and ultraviolet spectrum in this paper. In Gaussian software 09, the B3LYP density functional theory (DFT) was used to optimize the molecular geometric configuration at the base ground level of 6-311G (d,p). After frequency calculation, the infrared spectrum was obtained, and then the vibration characteristics were analyzed. It can be seen that the weight of all groups’ vibration in each vibration mode in the infrared spectrum of EGCG and GCG, and the corresponding vibration attribution and comparative analysis were made. It is found that the infrared spectra of molecule EGCG and GCG are similar. The absorption peaks for carbonyl stretching vibration are at 1 711 and 1 717 cm-1, respectively. The absorption peak of stretching vibration of phenolic hydroxyl groups on the benzene ring is mainly concentrated in 3 500~3 800 cm-1. Multiple absorption peaks in 1 000~1 600 cm-1 are in-plane bending vibration of benzene. The absorption peaks near 1 350 and 1 280 cm-1 are caused by methylene and methine in-plane bending vibration. The absorption peaks of out-plane bending vibrations are all below 500 cm-1. The infrared spectrum of EGCG molecule (400~4 000 cm-1) was measured through solid powder tableting method by using the IRPRESTIGE-21 infrared spectrometer manufactured by Shimadzu Corporation of Japan. The experimental infrared spectrum of EGCG is compared with the theoretical infrared spectrum. The result shows that the IR spectrum measured in the solid phase is almost consistent with the values calculated in gas phase. The theoretical infrared spectrum has slightly red-shift. The reason may be that the potential function used in theoretical calculation under the gas phase exist error. Compared with the gas phase without molecular interaction, the actual bond strength in the solid phase is slightly higher than that under the gas phase condition. In Gaussian software 09, the time-dependent density functional theory (TD-DFT) was used to calculate 15 excited states of EGCG molecules in ethanol solvent. The composition and energy level transition of the excited state were analyzed. The two absorption peaks by theoretical calculation were 229.3 and 276.4 nm, respectively. They were main corresponding the transition of p-π conjugated electron of p-electron and benzene ring π bond and the π-π* transition on benzene ring and heterocyclic ring. According to the analysis of the intensity of the oscillator, the transition from the ground state to S4, S5, S6 and S12 excited states is the main reason for the ultraviolet spectrum. The other excited state may be the forbidden transition, because the intensity of the oscillators are all less than 0.01. The above calculated value is almost consistent with the maximum absorption peak of the experimental values of EGCG. The absorption peak of experiment is at 235.1 and 278.7 nm in ethanol solvent. The calculated value is slightly blue-shift, which may be caused by the weak alkaline of the tea polyphenols or the weak alkaline of the molecules themselves. This study can provide theoretical reference for studying the properties and biological activities of EGCG and GCG molecules and the antioxidant properties of tea polyphenols.
现阶段, 茶叶已经在30多个国家种植, 主要集中在亚洲、 非洲和拉丁美洲。 其中, 中国、 印度、 斯里兰卡、 印尼、 肯尼亚、 土耳其的茶叶总产量占到全球产量的80%以上。 流行病学观察和实验室研究表明, 茶中存在的多酚类化合物是天然抗氧化剂, 可以抑制细胞衰老[1], 降低多种疾病的风险[2], 包括多种癌症、 动脉硬化、 冠心病等[3], 这也是茶叶广为流传的原因。 本文主要涉及绿茶。 绿茶采摘后主要通过晾晒、 杀青、 搓揉、 烘干等步骤完成, 与红茶、 黑茶等相比, 最大程度上保存了新鲜茶叶中的有机活性成分。 茶文化在中国更是历史悠久, 早有“ 宁可三日无米, 不可一日无茶” 的说法便说明了中国人一直以来对茶这种特别而又健康的饮料的钟爱。
绿茶中最主要的有机活性成分是茶多酚, 占茶叶干重的15%左右, 是茶叶抗氧化性的主体, 其中表没食子儿茶素没食子酸酯(EGCG)含量最高, 一般占茶多酚的40%以上[4], 它有一种异构体没食子儿茶素没食子酸酯(GCG)也主要存在于茶多酚中, 但含量较低。 冯国栋[5]等做了茶叶品种及采摘时节对其活性成分的影响分析, 余涛[6]等做了茶叶光谱与叶绿素、 茶氨酸、 茶多酚含量关系分析, 但没有分析茶多酚中的具体成分。 苏怡[7]等使用量子化学[8]的方法考察茶多酚中羟基的抗氧化活性; 王川丕[9]等用密度泛函理论研究了本文中相同的儿茶素EGCG与其空间异构体GCG的抗氧化活性和反应活性。 以上对茶叶性能的分析或基于部分实验或基于简单的理论计算, 没有对茶叶中最大活性成分茶多酚的物理化学性质进行更详细的分析。
为更好的分析茶多酚的性质, 本文针对茶多酚中EGCG和GCG两个分子, 一方面采用量子化学中密度泛函理论计算得到了它们的红外光谱和紫外光谱, 另一方面, 通过实验测量EGCG红外光谱及紫外光谱。 然后, 从理论上分析了其红外光谱对应的振动归属及紫外光谱对应的电子能级跃迁情况, 并将理论计算结果与实验结果进行比较, 发现它们吻合较好, 表明文中采用的计算基组及模拟方法是可行的。 本研究可加深人们对茶多酚中EGCG和GCG分子的认知, 以便今后更科学地运用茶多酚的功效。
本文光谱计算使用Gaussian 09软件的D01版本, 辅助以Gaussian view 6.0建模。 在气相环境下, 用B3LYP密度泛函的方法在6-311g(d, p)的基组水平上分别对表没食子儿茶素没食子酸酯(EGCG)和没食子儿茶素没食子酸酯(GCG)进行几何优化和频率计算(opt+freq), 结果均收敛, 无虚频且达到能量极小点, 得到稳定的结构。 在优化完成的基础上, 用关键字freq=intmodes进行分子骨架振动分析, 用TDDFT方法计算了分子的15个激发态, 另外用Multiwfn[10]波函数分析软件计算处理得到分子的红外光谱图和紫外光谱图。
EGCG粉末样品来源于西安博联特化工有限公司(纯度≥ 98%), 光谱仪分别为日本岛津公司生产的IRPRESTIGE-21红外光谱仪和上海MAPAD公司生产的UV-6100S型紫外分光光度计[11], 红外光谱测量采用粉末压片法(将烘干了的KBr和EGCG分子粉末混合后研磨均匀再压片), 紫外光谱测量采用液相法(以乙醇为溶剂)。
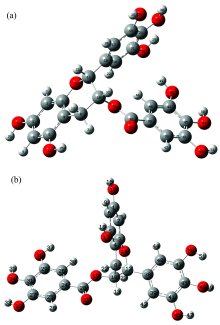
EGCG分子与GCG分子优化后的稳定几何构型分别如图1(a)和(b)所示, EGCG和GCG分子都有三个苯环, 一个六元杂环, 一个酯基。 为了方便称呼, 文中把连有两个羟基的苯环称为1号苯环, 把与六元杂环的一个C原子单键相连的称为2号苯环, 把连着C═O支链的苯环称为3号苯环。 由于分子没有对称性, 各个原子间作用力不同, 导致整个分子被扭曲, 六元杂环也并不共面, 成较复杂的空间立体结构。
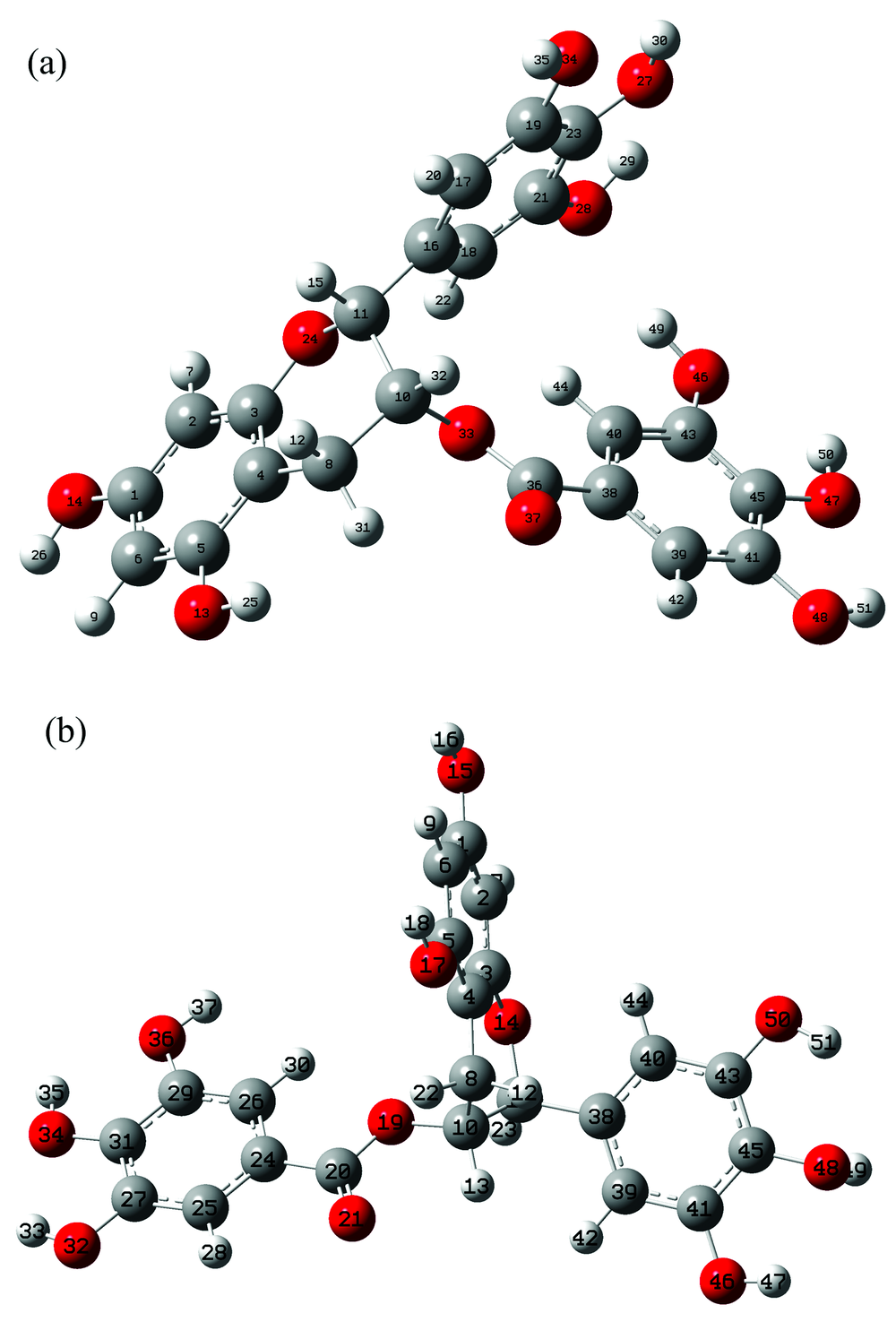 | 图1 分子构型 (a): 表没食子儿茶素没食子酸酯; (b): 没食子儿茶素没食子酸酯Fig.1 Molecular configuration (a): Epigallocatechin gallate; (b) Gallocatechin gallate |
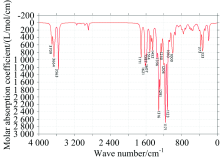
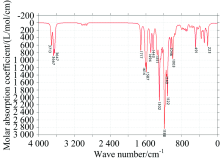
中红外光谱图可以反映分子转动和振动的特征, 特别是部分基团的性质, 利用红外光谱图都可以进行定性研究。 将Gaussian09软件计算的频率数据, 经Multiwfn软件进行频率校正因子(0.967)修正, 得到EGCG(图2)和GCG(图3)分子气相下的红外光谱图。
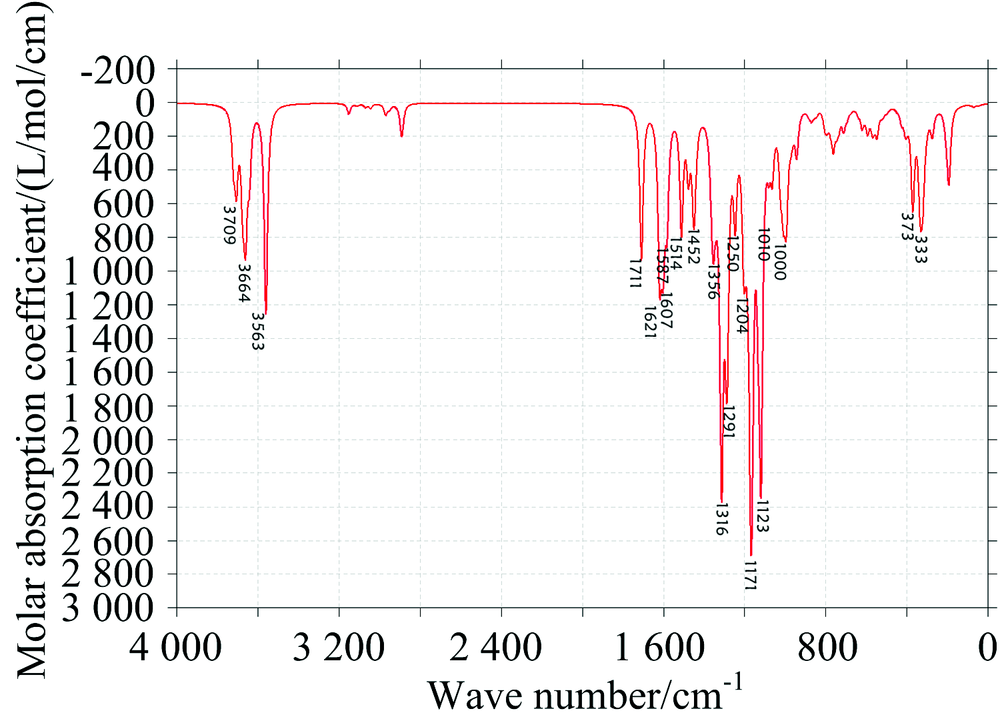 | 图2 EGCG的IR理论光谱图Fig.2 Theoretical IR spectrum of EGCG |
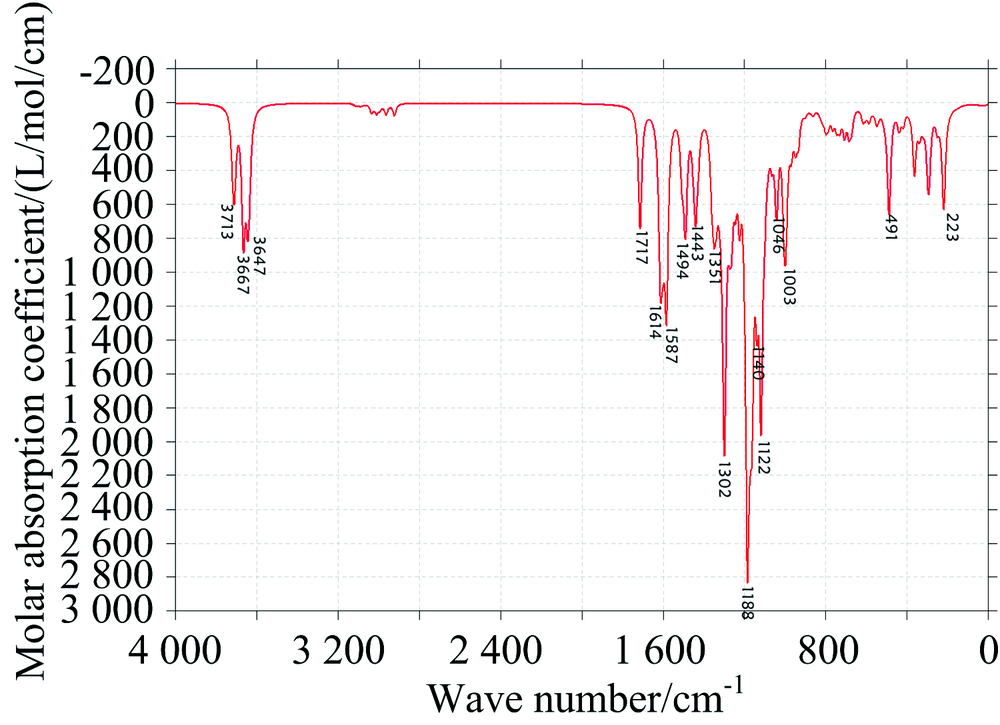 | 图3 GCG的IR理论光谱图Fig.3 Theoretical IR spectrum of GCG |
表1给出了计算红外谱图中EGCG分子较高的吸收峰的振动归属分析, EGCG与GCG两分子结构官能团都相同, 只有部分结构的相对位置略微不同, 故所得的IR谱图类似: 两分子苯环上酚羟基的伸缩振动都主要集中在3 500~3 800 cm-1, 靠近分子的羟基振动会受到分子间作用力的影响产生一定的偏移, 导致增强或减弱; EGCG的C═O伸缩振动位于1 711 cm-1处, GCG的位于1 717 cm-1处; 因为有3个苯环, 1 000~1 600 cm-1范围的每个峰都有苯环面内振动参与; 在1 356和1 291cm-1附近EGCG的亚甲基次甲基振动明显, 而GCG中亚甲基在其面内振动主要位于1 443 cm-1, 次甲基振动主要在1 272和1 248 cm-1附近; 在500 cm-1以下吸收峰大都为边缘处原子的面外振动。
| 表1 EGCG振动归属 Table 1 Assigned of vibration of EGCG |
特别指出的GCG的1 272 cm-1峰, 当苯环上的酚羟基如果不与苯环在同一平面上时, 苯环在面内振动带动羟基振动时也同时会造成苯环的二面角的改变, 使苯环上6个C原子偏离平面, 这里面外的H导致苯环二面角的改变振动占2%左右。 位于1180 cm-1附近的吸收峰最强, 是由于多原子振动的耦合产生的。
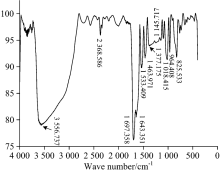
图4是实验测得的EGCG分子的IR光谱图, 对比理论计算的光谱图(图2), 发现理论计算数据与实验数据吻合较好。 在实验谱图中, 酚羟基的伸缩振动峰是宽峰, 最大峰值位于3 556.7 cm-1处对应计算谱图中的多个峰; 在2 368 cm-1附近的吸收峰为空气中测量光谱时CO2的吸收峰, 1 697 .4 cm-1峰对应理论计算图中1 711 cm-1; 1 643 cm-1对应计算图中1 621 cm-1; 1 533 .4cm-1对应1 514 cm-1, 1 463 cm-1对应1 452 cm-1, 1 377~1 145 cm-1宽峰对应计算谱图中多个苯环振动峰的耦合, 1 018 cm-1对应1 000 cm-1都对应较好。 相对于实际测量的谱图, 理论计算的谱图有轻微红移, 原因可能是计算在气相条件下采用的势函数存在误差, 实际测量的固相光谱的分子键强度比气相条件下略大些。
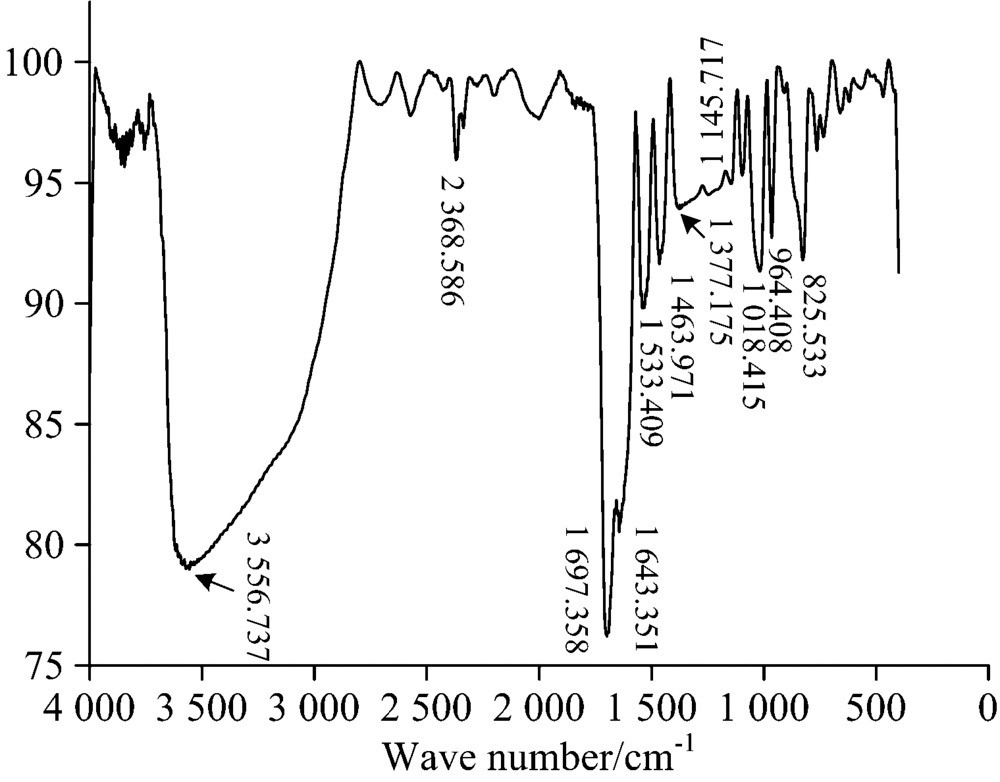 | 图4 EGCG的IR实验光谱图Fig.4 Experimental IR spectrum of EGCG |
分子的价电子吸收能量以后, 会跃迁到高能级, 所产生的吸收光谱波段一般在紫外到可见波段, 也被称为电子光谱, 本文中主要研究EGCG分子在200~350 nm波段的光谱。
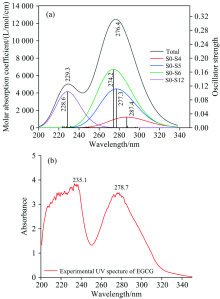
采用TD-DFT方法, 在结构优化的基础上计算了EGCG分子的15个激发态, 并得到了相应的UV光谱[图5(a)]。 图中S0表示基态, Sx表示第x个激发态。 计算所得2个吸收峰分别位于229.3和276.4 nm处, 主要分别对应p电子与苯环上π 键电子形成的p— π 共轭的电子跃迁及苯环、 杂环上π → π * 跃迁: 229.3 nm处的峰包含了1% 的S0— S6跃迁、 82.5%的S0— S12跃迁的贡献, 276.4 nm处吸收峰包含了8.16%的S0— S4跃迁、 35.77%的S0— S5跃迁和53.43%的S0— S6跃迁的贡献。 对这几个贡献较大的激发态进行激发性质的分析, 见表2。
| 表2 EGCG激发性质 Table 2 Excited state of EGCG |
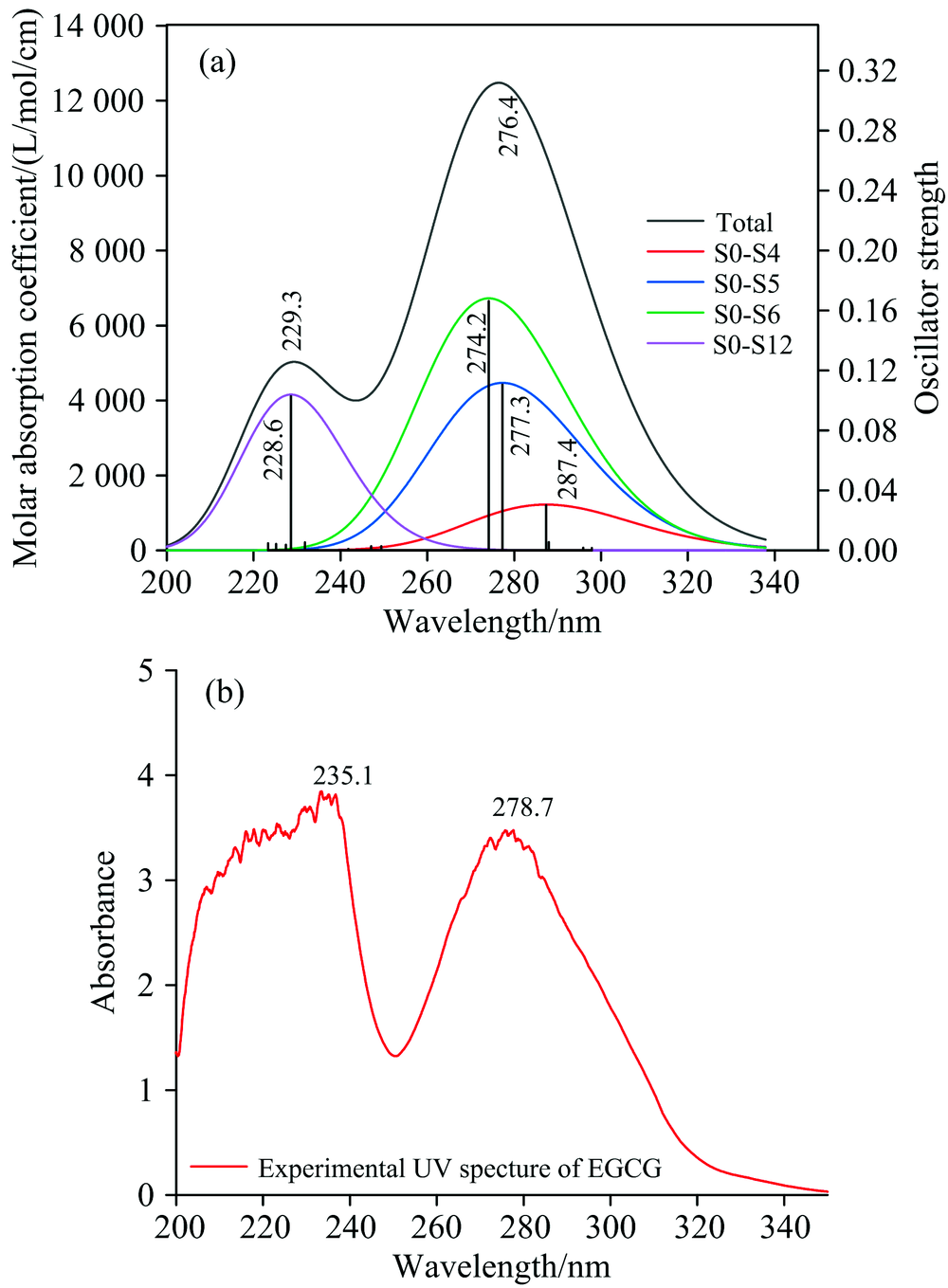 | 图5 EGCG分子紫外光谱 (a): EGCG的UV计算光谱图; (b): EGCG的UV实验光谱图Fig.5 The ultraviolet spectrum of EGCG molecules (a): Theoretical UV spectrum of EGCG; (b): Experimental UV spectrum of EGCG |
计算的15个激发态中, 另外几个激发态的振子强度均低于所设阈值0.01, 故不标出。
实验测量的EGCG样品的紫外光谱如图5(b)所示, 发现其吸收峰位置出现在235.1和278.7 nm处, 与理论计算值相比出现了整体轻微红移, 其原因可能是实验选用的茶多酚本身就具有的弱碱性或是在弱碱性环境下提取的, 使得溶液的PH值升高, 从而导致紫外吸收峰的整体红移。
本文中理论计算和实验测得的EGCG分子的紫外光谱与文献[12]中最大吸收峰274 nm测量值吻合较好, 误差产生的原因可能是使用的溶剂不同或药品来源及纯度不同而导致的。
(1)优化结果表明: EGCG与GCG分子都成较复杂的空间立体结构而非平面或对称结构。
(2)分析计算得到的IR谱图EGCG与GCG计算谱图大体一致, 苯环上酚羟基的伸缩振动主要集中在3 500~3 800 cm-1, C═O伸缩振动分别位于1 711和1 717 cm-1处, 因为有3个苯环, 1 000~1 600 cm-1的多个峰都有苯环面内振动参与, 在1 350和1 280 cm-1附近亚甲基次甲基振动明显, 在500 cm-1以下大都为原子的面外振动。 计算值与实验值吻合较好, 计算得到的谱图整体略微红移, 表明实际固相条件下, 由于分子间作用、 外界干扰等多方面原因, 分子键的强度要更大些。
(3)紫外光谱图中, 2个吸收峰229.3和276.4 nm主要对应苯环上p-π 键共轭的电子跃迁及杂环上π → π * 跃迁。 分析振子强度得知, 基态跃迁到S4, S5, S6和S12激发态为紫外光谱产生的主要原因。 分析这四个激发态的性质, 可以看到大多是由在HOMO和LUMO较近轨道的跃迁产生的。 其余的激发态可能为禁阻跃迁, 振子强度均小于0.01。 光谱的计算值与实验值和文献值均吻合较好, 实验值较计算值略有红移, 可能是因为分子本身的弱碱性条件所致。 本文实验值与文献实验值误差产生的原因可能是使用的溶剂不同或药品来源不同而导致的。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|