

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
利用太赫兹光谱检测萘啶酸的同质多晶
[肖田田 , 张卓勇, 郭长彬, 王果
, 张卓勇, 郭长彬, 王果* , 廖奕* ]
, 张卓勇, 郭长彬, 王果, 廖奕]
|
|


作者简介: 肖田田, 1992年生, 首都师范大学化学系硕士研究生 e-mail: xiaotian_dream@163.com
太赫兹光谱可以反映与固体中晶格振动有关的信息, 特别适合于检测相同分子形成的不同晶体, 因此利用太赫兹光谱对检测并控制药物的不同晶型具有重要的意义。 而药物分子的晶型会影响药物在存储中的稳定性等性质。 选取最早使用的喹诺酮类抗生素萘啶酸作为研究对象, 首先合成了萘啶酸的两种晶型NA-Ⅰ和NA-Ⅱ。 通过X射线粉末衍射确认了两种晶型。 使用太赫兹时域光谱技术在室温下对合成的两种萘啶酸不同晶型NA-Ⅰ和NA-Ⅱ在0.2~2.4 THz范围内进行检测, NA-Ⅰ和NA-Ⅱ在太赫兹光谱中表现出明显的差异。 NA-Ⅰ在0.94, 1.41, 1.88, 2.05和2.17THz处有五处特征峰, 而NA-Ⅱ在0.72, 0.96, 1.38, 1.80, 2.04和2.16 THz处有六处特征峰。 二者最明显的差异是NA-Ⅱ在2.04 THz处的峰为肩峰, 而NA-Ⅰ在2.05 THz处的吸收强度要大得多。 不同于其他光谱, 如红外光谱, 太赫兹光谱中的特征峰不对应特定的官能团, 因此对光谱峰的指认尤为重要。 利用密度泛函理论对两种晶型进行理论计算, 理论峰与实验特征峰基本对应, 得出萘啶酸两种晶型在此低频范围的振动模式主要是分子的骨架运动, 并从两种晶体堆积方式的不同阐述了光谱显著差异的原因。 该研究为利用太赫兹光谱检测药物的同质多晶提供了依据。
Terahertz spectroscopy can provide the lattice vibration information of solid materials, which is different from previous measuring methods. And particularly, it is suitable to test polymorphs. Thus it has an important meaning for controlling and detecting the polymorphs of drugs. The polymorphs should affect the properties, such as stability,of the drugs in storage. Nalidixic acid, the earliest quinolone antibacterials used, was investigated in this work. Two polymorphs NA-Ⅰ and NA-Ⅱ were synthesized and were confirmed by X-ray powder diffraction experiments. Different absorption spectra in the range of 0.2~2.4 THz of two synthesized polymorphs of nalidixic acid were obtained with terahertz time-domain spectroscopy at room temperature. Significant differences existed in the terahertz spectra of NA-Ⅰ and NA-Ⅱ. Five characteristic absorption peaks observed in the terahertz spectrum of NA-Ⅰ were at 0.94, 1.41, 1.88, 2.05, and 2.16 THz, while six peaks of NA-Ⅱ were at 0.72, 0.96, 1.38, 1.80, 2.04 and 2.16 THz. The most obvious difference was that there was a shoulder peak at 2.04 THz for NA-Ⅱ, while the intensity of the peak at 2.05 THz for NA-Ⅱ was much larger. Unlike other spectroscopies, such as infrared spectroscopy, the characteristic peaks in terahertz spectra were not related to specific functional groups. Thus the assignment of the characteristic peaks in terahertz spectra was essential. Theoretical calculations for the two polymorphs were performed based on density functional theory. The calculated characteristic peaks were in accord with those in the experiments. The vibrational modes in this low frequency region were mainly skeletal vibrations. Also the difference of the two spectra was explained by the different packing modes of the two crystals. This work provides evidence for identification of drug polymorphs using terahertz spectroscopy.
同质多晶是指同一种化合物由于在晶体中排列方式的不同而存在两种或多种晶体结构的现象[1, 2]。 超过50%的药物活性成分具有同质多晶现象[3]。 因为不同晶型的药物分子具有不同的物理、 化学性质, 如熔点、 密度、 稳定性、 加工性、 溶出速率和生物利用度等[4], 在制药行业中检测与控制药物化合物的晶型是确保药物质量和药物疗效的一个重要环节。
近年来, 鉴别不同药物晶型之间的差异成为研究热点。 表征药物不同晶型的技术有许多, 如X射线粉末衍射、 红外、 拉曼光谱以及热重分析等。 但是上述技术手段都有不同程度的局限, 如X射线粉末衍射存在的电离效应, 如何安全应用还有待考量; 红外、 拉曼光谱对于具有相同官能团的分子区分不太好; 热重分析对于熔点接近的不同晶型不能得到准确可靠的结果。 而太赫兹时域光谱(Terahertz time-domain spectroscopy, THz-TDS)的频率范围在0.1~10 THz, 其光子能量与大部分有机分子及分子团的振动和转动能级之间跃迁的能量相当, 包含丰富的物理和化学信息, 能够对材料组成及结构的细微变化做出分析和鉴定, 从而被广泛应用于药物和生物领域, THz-TDS正在成为安全方便地检测药物不同晶型的新方法[5]。 然而太赫兹光谱不像红外光谱那样, 一定位置的峰对应相应官能团的振动。 因此如何指认谱峰、 解释不同晶型谱峰差异的来源成为一个具有挑战性的工作。 太赫兹波段的能量对应固体中分子间弱的相互作用(如氢键、 范德华作用等)、 大分子的骨架振动以及晶体中晶格的低频振动等[5, 6]。 由于药物分子在固体状态时具有分子间相互作用, 而单分子模型未包含这类相互作用, 因此必须采用分子簇模型[7]甚至是周期性结构模型[8, 9]才能较好地描述这些体系。
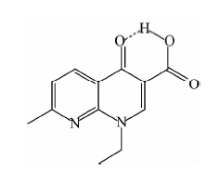
萘啶酸(nalidixic acid, NA), 分子结构见图1, 作为一种药物, 是最早使用的喹诺酮类抗生素之一, 能够有效的抑制革兰氏阳性菌和革兰氏阴性菌, 它主要用于治疗尿路感染, 是美国食品药品监督管理局唯一批准用于儿科的喹诺酮药物[10]。 近年来研究发现, NA具有三种不同的晶型, 相比于其他两种晶型, NA-Ⅲ 在室温下不能稳定存在[10]。 由于NA是一种药物, 从药物检测的实用角度出发, 本研究并未考虑NA-Ⅲ 。 区分并鉴别其他两种晶型NA-Ⅰ 和NA-Ⅱ , 对于深入研究其理化性质以及药物监测管理有重大的理论和实际意义。 在本文中, 我们在室温下利用THz-TDS对NA的多晶粉末进行了测量, 获得了NA-Ⅰ 和NA-Ⅱ 在0.1~2.4 THz范围内的太赫兹吸收谱线, 两种晶型的谱图有明显的不同, 这表明THz-TDS能够为作为一种实验手段去检测不同晶型的NA药物分子。 基于密度泛函理论对NA-Ⅰ 和NA-Ⅱ 晶体的太赫兹吸收光谱进行了计算, 对实验中的吸收峰进行了指认, 从晶体的堆积方式出发, 阐述了两种晶型谱峰差异性的来源。
 | 图1 NA的分子结构Fig.1 The structure of NA |
1.1.1 样品制备
原材料萘啶酸(纯度大于95.0%(T, 滴定法测试))从梯希爱(TCI)(上海)化成工业有限公司购买, 使用前没有经过进一步纯化。 将萘啶酸原材料溶解于甲醇(AR, 分析纯)中, 在室温下缓慢蒸发得到平行四边形的NA-Ⅰ 晶体。 称取100 mg萘啶酸, 加入到10 mL的二氯甲烷(AR, 分析纯)中, 加热并使用超声使其完全溶解, 冷却至室温, 加入40 mL正己烷(AR, 分析纯)作为抗溶剂, 静置, 待晶体完全析出后过滤、 干燥, 得到棍状晶体NA-Ⅱ [10]。
1.1.2 X射线粉末衍射
为了验证得到的样品是否是预期的晶体, 使用BrukerD8 advance X射线粉末衍射仪, 采用Cu靶产生的Cu-Kα 射线对晶体粉末进行扫描, 操作电压和电流分别是30 kV和30 mA, 在2θ 为10° ~30° 的范围内收集数据, 数据用MDI JADE程序进行处理。
1.1.3 THz-TDS
利用太赫兹时域系统进行光谱测试, 产生太赫兹脉冲的频率范围约为0.2~3 THz。 实验中用NA-Ⅰ 和NA-Ⅱ 各40 mg分别与80 mg聚乙烯粉末(在太赫兹波段几乎透明)混合, 将混合物研磨以获得均匀粉末, 粒度达到几微米以减少散射效应。 将混合物倒入钢模在7 t压片5 min, 得到直径为13 mm的无裂缝且两端面平行的样片。 厚度测量利用电子游标卡尺, 每个样品选择不同的三个点, 并求取平均值, 平均厚度在1 mm左右。 太赫兹光路和样品罩在充有氮气的箱体内, 以减少空气中水分对太赫兹波的吸收并提高信噪比, 箱内的相对湿度小于1%, 温度为室温。 NA-Ⅰ 和NA-Ⅱ 各压两个平行片, 每个样品正反各测一次, 并对测得的光谱求平均值。
如图1所示, NA分子中羧基上的氢和羰基可以形成分子内氢键, 这一点在晶体中仍然存在[10]。 如图2所示, 萘啶酸分子除了甲基和乙基外, 原子基本处于同一个平面。 NA-Ⅰ 和NA-Ⅱ 都为单斜晶体[10], 不同的是: 在NA-Ⅱ 中分子基本靠π — π 堆积方式构成晶体; 而在NA-Ⅰ 中, 除了π — π 堆积外, 分子间还存在边对面堆积方式。 堆积方式的不同可能会导致不同的光谱性质。 在图3中, NA-Ⅰ 和NA-Ⅱ 的X射线粉末衍射峰是不同的, 为了鉴别两种实验中获得的晶型, 使用Mercury程序从单晶结构出发模拟了NA-Ⅰ 和NA-Ⅱ 的衍射峰, 通过对比发现, 实验峰和理论模拟峰基本对应,
 | 图2 NA-Ⅰ (a)和NA-Ⅱ (b)的晶体结构Fig.2 The crystal structure of NA-Ⅰ (a) and NA-Ⅱ (b) |
其中10° ~12° 处的峰与理论稍有差异, 是由于XRD多晶粉末衍射制样晶粒大小改变, 会导致峰位偏移, 但特征峰与理论峰对应, 推断实验上获得的晶体确为NA-Ⅰ 和NA-Ⅱ 。 NA-Ⅰ 和NA-Ⅱ 在2.4 THz以下的太赫兹光谱如图4所示。 两种堆积方式不同的晶体显示出不同的太赫兹光谱。 NA-Ⅰ 在0.94, 1.41, 1.88, 2.05和2.17 THz处存在五处特征吸收峰, 而NA-Ⅱ 在0.72, 0.96, 1.38, 1.80, 2.04和2.16 THz处存在六处特征吸收峰。 NA-Ⅱ 比NA-Ⅰ 多了一个在0.72 THz处吸收强度较小的吸收峰, 其他相应峰的位置比较接近。 两幅太赫兹光谱最明显的不同在于NA-Ⅰ 在2.05 THz处的吸收强度比NA-Ⅱ 在2.04 THz处的大很多。 通过该处吸收峰与2.17或2.16 THz处吸收强度的相对大小可以判断两种晶型: NA-Ⅰ 在这两处形成双峰, 而NA-Ⅱ 只在单峰旁有一个肩峰, 通过此处可以鉴别NA-Ⅰ 和NA-Ⅱ 。
 | 图3 NA-Ⅰ (a)和 NA-Ⅱ (b)的X射线粉末衍射实验和理论模拟图Fig.3 Experimental and simulated X-ray powder diffraction patterns of NA-Ⅰ (a) and NA-Ⅱ (b) |
 | 图4 NA-Ⅰ (a)和 NA-Ⅱ (b)太赫兹实验和理论模拟图Fig.4 Experimental and calculated terahertz spectra of NA-Ⅰ (a) and NA-Ⅱ (b) |
通过理论计算得到的NA-Ⅰ 和NA-Ⅱ 的太赫兹光谱也如图4所示。 将理论计算得到的特征吸收峰拟合后得到平滑曲线后, 峰的数目与实验得到的一致, 其对应关系如表1中所列。 如图4和表1所示, 理论计算所得到的峰的位置比实验的峰普遍偏高, 部分原因是通过密度泛函理论计算的频率对应绝对零度下的数值, 而实验测量由于实际应用方面的原因是在常温下进行的, 有文献表明太赫兹光谱存在温度效应[16], 即实验测量的谱峰随温度的降低而升高, 温度效应部分解释了理论计算得到的数值偏高的原因, 幸运的是峰位置的平移是整体的, 不影响各峰的相对位置, 因此不影响理论计算对实验峰的指认。 还有部分原因是采用的optB88-vdW密度泛函和PAW基函数本身的误差。 虽然有频率校正因子, 但是针对振动频率的校正多基于Gauss型基函数, 因此本研究未使用校正因子。 为了检验optB88-vdW密度泛函和PAW基函数的误差, 我们需要用一个模型分子作为验证。 由于萘啶酸属于芳香类有机化合物, 我们选取了最简单的芳香类有机化合物— — 苯分子进行了振动分析, 用optB88-vdW密度泛函得到的三个最低振动频率为412, 601和700 cm-1(前两个振动模式是二重简并的), 它们比用常见PBE密度泛函计算出来的399, 600和667 cm-1要高。 对于不同的振动模式, 高出的数值也不同, 其中第三个频率高出33 cm-1。 由此可见, 使用optB88-vdW密度泛函和PAW基函数计算出来的振动频率确实会偏高。 因此采用该方法计算的萘啶酸谱峰数值偏高这一点不影响对实验峰的指认。
| 表1 NA-Ⅰ 和NA-Ⅱ 的实验特征峰和理论计算特征峰以及对特征峰的归属 Table 1 Assignments of the absorption peaks for NA-Ⅰ and NA-Ⅱ |
如表1所列, NA-Ⅰ 的第3, 4, 5处实验中吸收峰的吸收强度依次递增, 而理论计算的依次递减, 但都是吸收强度较大的峰, 不影响指认, 其余峰的相对吸收强度是一致的; 理论计算的NA-Ⅱ 各峰的吸收强度与实验基本一致, 第5处峰的吸收强度较小, 实验中也是这样。 理论计算获得的NA-Ⅰ 的第4个峰的相对吸收强度明显比NA-Ⅱ 的第5个峰大, 这一点和前述实验中用于鉴别NA-Ⅰ 和NA-Ⅱ 吸收峰的关键区别相一致。
如图4所示, 在理论计算中存在个别吸收峰由不止一个振动模式合并产生, 在对实验峰的指认中, 只在表1中列出了吸收强度最大的振动模式。 将VASP程序的振动分析结果处理后, 用GaussView程序将振动模式图形化, 如图5、 图6所示。 表1中NA-Ⅰ 第1个吸收峰对应的振动模式是边对面分子之间距离的变化, 如前所述, NA-Ⅰ 中存在π -π 堆积和边对面两种分子堆积方式, 在该振动模式中, 以π — π 堆积方式结合的分子朝相同的方向运动, 不存在相对位移, 而不同朝向的π — π 堆积而成的分子之间存在相对位移, 这对应边和面之间距离的变化。 NA-Ⅰ 第2个吸收峰对应π — π 堆积分子之间的相对扭转, 以π — π 堆积方式结合的相邻层分子的转动方向是相反的, 这就造成了π — π 堆积分子相邻层之间的相对扭转, 以边和面方式结合的分子之间的位置变化不明显。 在NA-Ⅰ 第3个吸收峰处, 除了与第2个吸收峰相同的运动外, 还有甲基的扭转。 在NA-Ⅰ 第4个吸收峰处, 除了甲基的扭转外, 还有羧基(特别是羧基中的羰基氧)偏离分子平面的摇摆振动。 在NA-Ⅰ 第5个吸收峰处, 除了甲基的扭转外, 还有分子平面的面外变形振动, 具体的是分子两端羧基和甲基朝垂直于环的一面运动, 而分子中部朝相反的方向运动, 所有以π — π 堆积方式结合的分子的运动方向是一致的。 对于NA-Ⅱ 而言, 在表1中第1个吸收峰处, 以π — π 堆积方式结合的分子之间没有相对运动, 而整个堆积层绕垂直于分子平面的轴旋转, 相邻的不同堆积层的旋转方向相反, 造成了π — π 堆积层之间的面内相对扭转。 在第2个吸收峰处, 相邻的π — π 堆积层沿平行于分子平面的轴沿相反的方向旋转, 造成不同堆积层之间夹角的变化, 与第1个吸收峰不同, 分子偏离了原来的平面, 称为π — π 堆积层之间的面外相对扭转。 在第3个吸收峰处对应相邻层π — π 堆积分子之间的相对扭转。 在第4个吸收峰处除了甲基扭转外, 还有羧基(特别是羧基中的羰基氧)偏离分子平面的摇摆振动。 在第5个吸收峰处, 除了甲基、 乙基的扭转外, 还有相邻层π — π 堆积分子之间的相对平移。 在第6个吸收峰处, 除了甲基、 乙基的扭转外, 还有羧基偏离分子平面的摇摆振动。 NA-Ⅰ 和NA-Ⅱ 在该太赫兹能量范围内的运动除了羧基的面外摇摆振动、 甲基或乙基的扭转外, 主要是分子骨架的整体运动。 非谐效应常常发生在具有平缓势能面的骨架振动模式[17], 不同的振动模式所对应势能面的平缓程度可能不同, 因此造成的非谐效应的大小也可能有所不同, 这也是影响理论计算所得到峰位置偏移的部分原因。
 | 图5 NA-Ⅰ 的振动模式Fig.5 Vibrational modes of NA-Ⅰ |
 | 图6 NA-Ⅱ 的振动模式Fig.6 Vibrational modes of NA-Ⅱ |
在实验中两幅太赫兹光谱最明显的不同在于NA-Ⅱ 的第5个吸收峰的吸收强度较小。 而吸收强度与振动中偶极矩的变化有关, 在该吸收峰处, 甲基和乙基个别小基团的扭转对整体偶极矩的变化影响很小, 更为关键的是分子骨架的运动是π — π 堆积分子之间的相对平移, 由于分子是近平面的, 分子之间靠π — π 堆积这种弱的相互作用结合, 与其他类型的运动(如扭转)相比, 平动对偶极矩变化的贡献较小, 因此峰的吸收强度较小。 这种运动在NA-Ⅰ 中是不存在的, 因为在NA-Ⅰ 中, 除了π — π 堆积, 还有边对面堆积方式, 即使有这种类型的平动, 也会造成边与面距离的变化。 NA-Ⅱ 这种单一的堆积方式造成了它区别于NA-Ⅰ 的吸收峰, 从而被用于鉴别两种晶型。
合成了两种不同晶型的萘啶酸多晶粉末NA-Ⅰ 和NA-Ⅱ , 通过X射线粉末衍射方法验证了其结构。 用THz-TDS在室温下对两种不同晶型在0.2~2.4 THz范围内进行表征, 发现二者的吸收光谱具有明显的差异。 NA-Ⅰ 在0.94, 1.41, 1.88, 2.05和2.17 THz处有五处特征峰, 而NA-Ⅱ 在0.72, 0.96, 1.38, 1.80, 2.04和2.16 THz处有六处特征峰。 特别地, NA-Ⅱ 在2.01 THz处的吸收峰为肩峰, 而NA-Ⅰ 在此附近2.05 THz处峰的吸收强度明显大得多, 该处的显著区别可以用于两种晶型的鉴别。 利用密度泛函理论对NA-Ⅰ 和NA-Ⅱ 两种晶型进行理论研究, 对特征峰进行了归属和分析。 理论特征峰的数量和形状与实验特征峰能够基本对应, 峰频率上的误差来源于计算方法的误差、 温度效应以及非谐效应。 特别地, 实验中两种晶型的太赫兹吸收峰最显著的差异也被理论计算所验证, 并且从两种晶型堆积方式的角度阐述了光谱显著差异产生的原因。 通过比较实验和理论计算的结果, 得出萘啶酸两种晶型在此低频范围的振动模式除了羧基的面外摇摆振动、 甲基或乙基的扭转外, 主要是分子骨架的整体运动。 这一结果显示了太赫兹光谱技术在检测同质多晶中强大的能力, 为利用THz-TDS技术检测萘啶酸药物分子的同质多晶以及研究其分子间相互作用提供了实验依据和理论参考, 同时也证实了THz-TDS技术在药物多晶型安全检测方面的优越性。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|