

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
碱性介质电催化体系活性氧的生成及其光谱学研究
[薛玉冬1, 2  , 郑诗礼
, 郑诗礼1, * , 张懿1 , 金伟1, * ]
, 郑诗礼, 张懿]
|
|


作者简介: 薛玉冬, 1993年生, 中国科学院过程工程研究所博士研究生 e-mail: ydxueipe@hotmail.com
亚熔盐液相氧化技术可在相对低温下实现难分解两性金属矿物的高效转化, 基于此进一步提出了碱性介质电化学活性氧协同强化新方法。 利用紫外-可见光光谱和电子自旋共振波谱等手段对电催化体系活性氧的生成与转化机理进行了系统解析。 通过阴极电催化的作用, 在电极表面定向进行两电子氧气还原反应, 原位产生大量的活性氧组分。 研究发现过渡金属离子与两电子氧气还原产物H
Liquid oxidation techniques by sub-molten salt media for the effective extraction of amphoteric metal from refractory minerals have been developed. An innovative synergistic process by electrochemical oxidation and reactive oxygen species (ROS) intensify was further proposed. In the present study, the mechanism of ROS formation and conversion in the electrocatalytic process was systematically revealed by UV-Vis spectra and electron spin resonance (ESR) measurement. The reactive species were in-situ generated by two-electron oxygen reduction reaction (ORR) at the cathode surface. The ·OH was produced from the self-induced reaction between amphoteric metal ions and H
亚熔盐非常规介质可以实现难分解两性金属矿物的高效转化, 是解决我国大宗难处理两性金属矿物的化学冶金新途径。 基态氧分子可以在碱性介质中发生还原反应产生一系列具有强氧化性的活性氧中间产物, 包括· OH, ·
液相氧化反应体系已在铬、 钒等行业得以应用, 其中铬铁矿亚熔盐液相氧化清洁生产技术已被2016年环保部发布的《铬盐工业污染防治技术政策》鼓励采用; 亚熔盐法清洁提钒生产线也正式投入运营。 含铬或含钒的低价两性金属氧化物的氧化溶出是液相氧化反应体系中最重要的步骤。 本工作采用三氧化二铬和三氧化二钒的氧化溶出作为碱性介质电催化体系活性氧作用的体现, 对实际电化学清洁生产工艺提供理论依据。 通常的电化学高级氧化技术均限制在酸性或近中性的体系反应, 主要关注的是在污水处理、 有机物降解等方面的应用[3, 4], 而在碱性介质中对高级氧化技术的研究很少, 本研究通过调控碱性介质活性氧的组分实现碱性介质高级氧化, 并应用于矿物氧化溶出领域。
基于前期通过电化学方法揭示的碱性介质活性氧赋存和量化调控规律[1], 提出碱性介质电化学活性氧协同强化新方法, 利用“ 清洁高效” 的电能和氧气, 在阳极发生矿物直接氧化溶出, 而在阴极发生活性氧原位生成及其对矿物的间接氧化, 两者协同作用将反应条件拓展至更温和区域。 重点研究了通过阴极电催化电极的作用, 定向进行两电子氧气还原反应, 产生大量的活性氧组分, 促进两性金属的高效转化, 在温和区域内实现矿物溶出。 利用紫外-可见光光谱和电子自旋共振波谱等光谱学手段, 系统地揭示了反应机理。
高纯N2(≥ 99.999%)、 高纯O2(≥ 99.999%)购于北京千禧京城气体有限公司, Cr2O3(≥ 99.97%), V2O3(≥ 95%), KOH(≥ 85%)购于阿法埃莎(中国)有限公司, 5, 5-二甲基-1-吡咯啉-N-氧化物(DMPO, ≥ 97%), 二苯胺磺酸钠(≥ 97%)购于美国阿拉丁公司, H2O2(30%)、 C4K2O9Ti· 2H2O(≥ 98.5%)、 K2CrO7(≥ 99.8%)、 叔丁醇(≥ 98%)、 丙酮(≥ 99.5%)、 乙醇(≥ 95%)均为分析纯购于国药集团化学试剂北京有限公司, 超纯水(美国Millipore公司)。
上海辰华有限公司CHI760E电化学工作站, 美国Lab Tech公司UV9100紫外-可见光分光光度计, 日本岛津公司的JES-FA200电子自旋共振(ESR)波谱仪, 美国Perkin Elmer公司Optima 7300DV电感耦合等离子原子发射光谱仪(ICP-OES), 梅特勒-托利多仪器上海有限公司AE 163精密电子天平, 江苏金怡仪器科技有限公司85-1磁力搅拌器。
采用计时电流法作为电化学活性氧的生成与活性氧催化氧化反应手段[5]; 工作电极为碳毡电极(1 cm× 2 cm), 对电极为石墨电极(4 cm× 4 cm), 参比电极为汞-氧化汞电极; 设置的工作电位为阴极氧还原电位-1 V(vs. Hg/HgO)。 体系中加入的Cr2O3和V2O3均为过200目筛(< 75 μ m)的粉体。 实验主要通过紫外-可见光光谱在400 nm左右检测溶液中的H
以KOH溶液作为电解质, 将高纯氧气通入到400 mL电解槽中, 氧气会以溶解氧的形式存在并传递至阴极表面, 接着在阴极表面发生电化学还原反应。 在碱性介质中, 氧气可以进行四电子或二电子还原反应, 如式(1)和式(2)所示[8], 二电子氧还原反应产生的是H
为测定碱性介质电催化体系中H
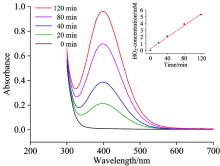
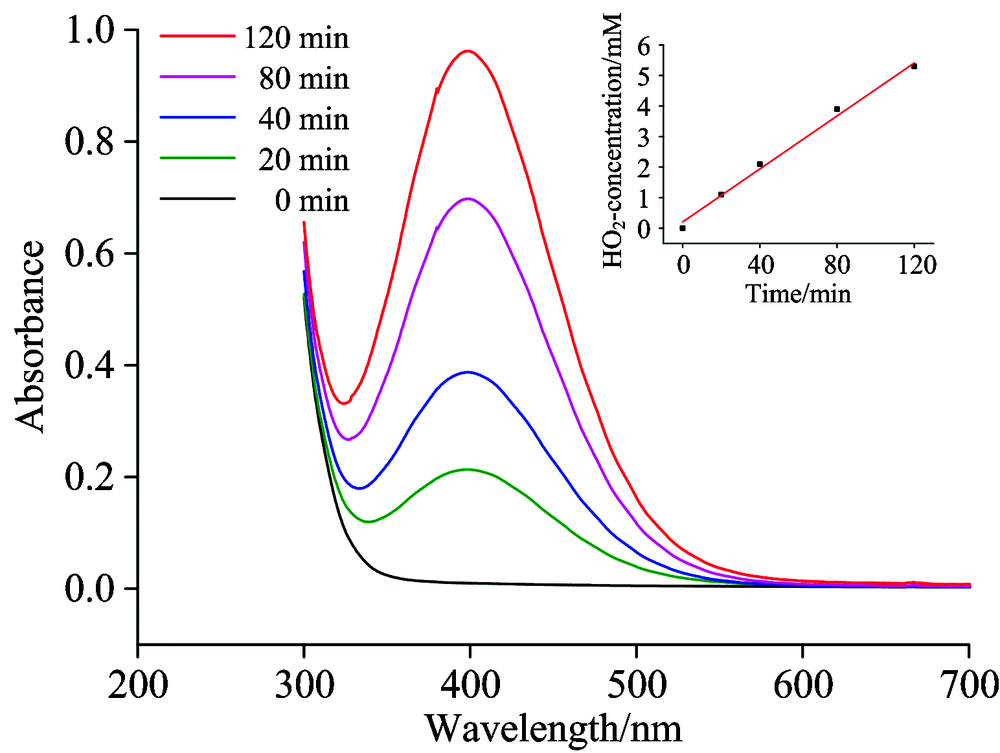 | 图1 紫外-可见光光谱测定电催化体系产生的H |
向KOH电解质中加入400 mg Cr2O3, 分别进行不同时间的电催化反应, 并用UV-Vis光谱对样品直接进行扫描, 结果分别如图2所示。 反应初始, 取样测试的结果表明300~500 nm范围内没有峰出现, 而反应60 min的取样结果表明, 360 nm处出现了谱峰, 此为碱性介质Cr(Ⅵ )的峰, 存在形式是Cr
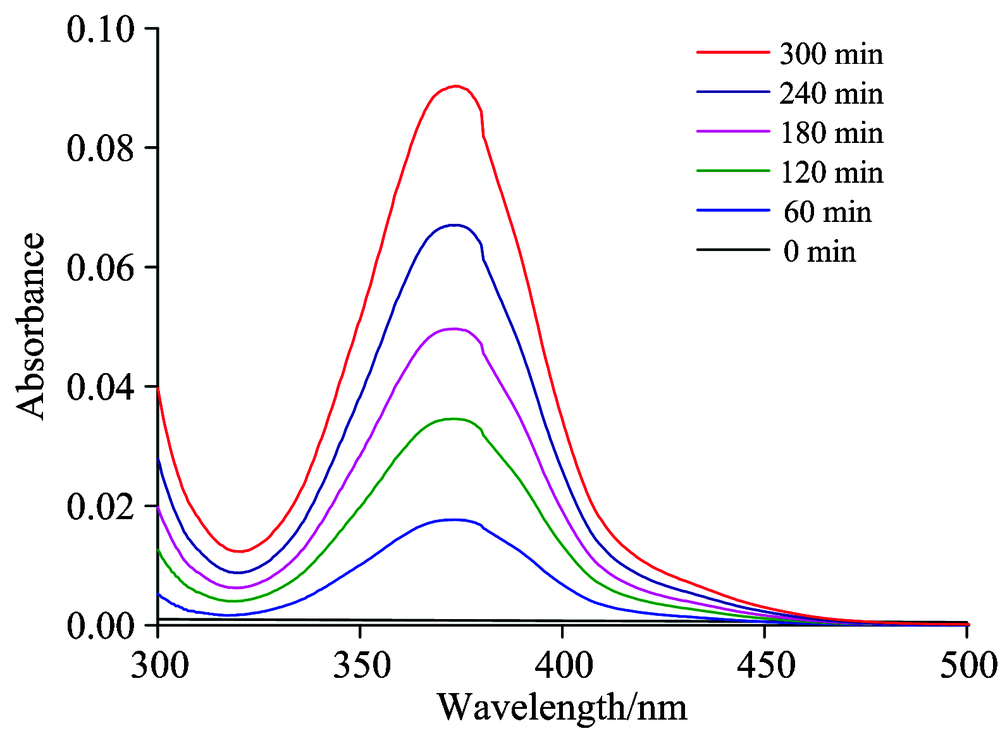 | 图2 紫外-可见光光谱测定电催化体系产生的Cr(Ⅵ )Fig.2 Chromium(Ⅵ ) ions generated from the electrocatalytic system determined by UV-Vis spectroscopy |
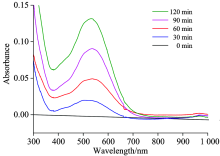
向KOH电解质中分别加入40 mg V2O3, 分别进行不同时间的电催化反应。 样品中V(Ⅴ )的测试原理是利用五价钒离子在强酸中有显著的氧化性, 能与二苯胺磺酸钠发生氧化还原显色反应, 可用分光光度法测定。 如图3所示, 测试结果表明在538 nm处出现了代表V(Ⅴ )的谱峰, 随着电催化反应的进行, 峰强度逐渐增加; 因此可以判断, V2O3在碱性介质活性氧的作用下同样实现了氧化溶出。
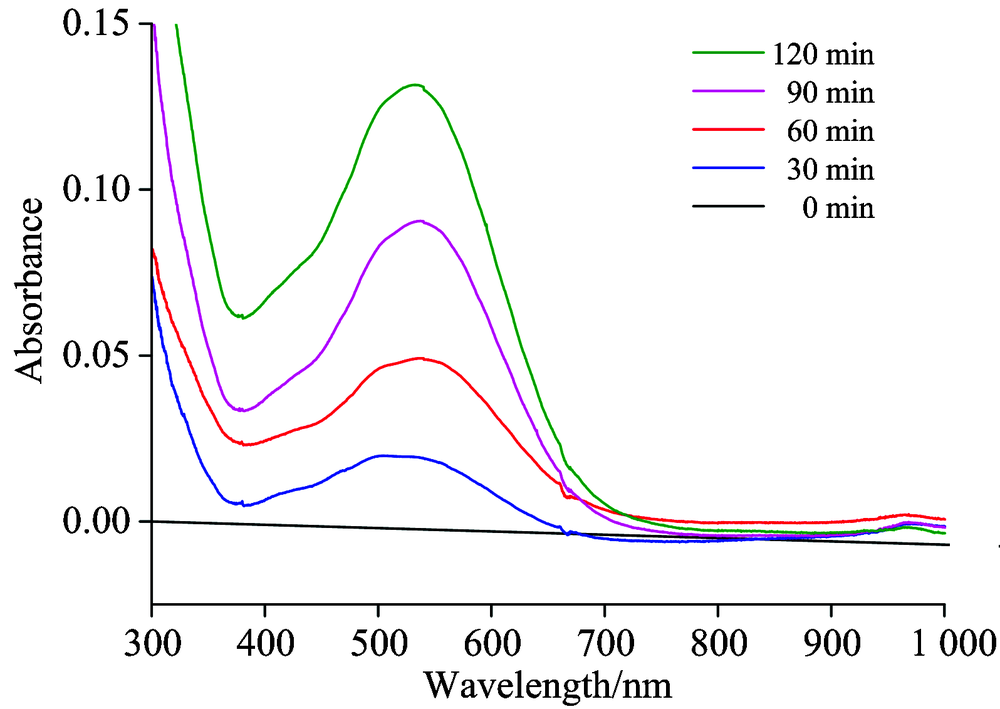 | 图3 紫外-可见光光谱测定电催化体系产生的V(Ⅴ )Fig.3 Vanadium(Ⅴ ) ions generated from the electrocatalytic system determined by UV-Vis spectroscopy |
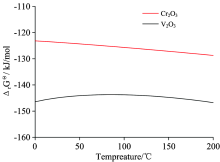
碱性介质电催化体系应用于V2O3和Cr2O3的氧化溶出过程, 是一种新型催化氧化反应体系, 为了更清楚了解氧化溶出过程的难易程度, 图4计算了反应(3)和(4)的标准自由能变化的热力学数据, 从图中可以看出, 在0~200 ℃的温度范围内, V2O3和Cr2O3氧化反应的吉布斯自由能均为负值, 且在此温度范围内V2O3氧化反应的吉布斯自由能均负于Cr2O3, 实验过程中采用的电催化体系为室温25 ℃左右, 二者的吉布斯自由能分别为-144.87和-123.75 kJ· mol-1, 表明了V2O3氧化溶出反应更易发生。
 | 图4 碱性介质中V2O3和Cr2O3氧化反应的Ellingham图Fig.4 Ellingham diagram for V2O3 and Cr2O3 oxidation reactions in alkaline media |
图5给出了DMPO作为羟基自由基捕获剂时, 不同体系的电子自旋共振(ESR)谱图。 由图可见, 在通入氧气的情况下, 活性氧催化氧化Cr2O3和V2O3的体系中得到了典型的羟基自由基1:2:2:1的信号峰, 证实了体系内羟基自由基的激发[9]。 在没有氧气的情况下, 只依靠电化学阳极氧化的作用, 体系内没有羟基自由基信号峰出现, 而且阳极采用石墨电极而非铂片电极, 避免了阳极直接产生羟基自由基的影响, 所以体系内没有检测到羟基自由基的谱峰。 此外, 在体系内不存在Cr和V的情况下, 单纯的二电子氧气还原反应体系下未能检测到羟基自由基的出现。
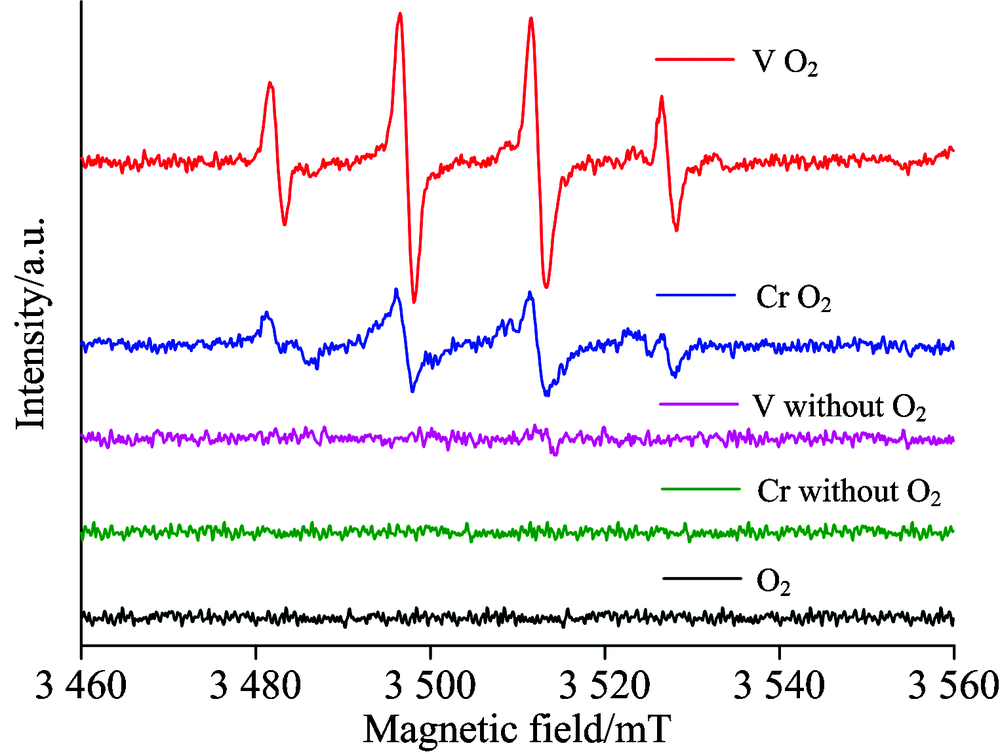 | 图5 电催化体系中利用DMPO作为羟基自由基捕获剂的电子自旋共振波谱图Fig.5 ESR spectroscopy for the hydroxyl radical detection in the typical electro-catalytic process with DMPO as a trapping agent |
电催化体系内羟基自由基的产生, 是由低价过渡金属氧化物中存在的低价过渡金属离子V(Ⅲ )和Cr(Ⅲ )在碱性介质活性氧H
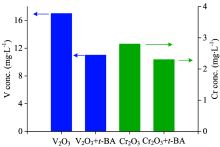
图6是利用ICP-OES对电催化体系钒和铬的溶出情况进行检测, 2 h钒溶出浓度大约是17 mg· L-1, 而铬溶出浓度相对较低, 只有2.8 mg· L-1, 此结果与热力学计算吻合, 证实了三氧化二铬更难被氧化。 还利用叔丁醇(t-BA)作为羟基自由基的捕获剂[11], 将其加入钒铬溶出体系中, 抑制羟基自由基的作用。 结果表明, 加入t-BA后, 钒和铬的溶出浓度均有不同程度的下降, 验证了钒、 铬自诱导过程中产生的羟基自由基可以作为活性氧组分, 继续促进氧化溶出反应的进行。
 | 图6 碱性介质2 h电催化反应后体系内的钒、 铬溶出浓度Fig.6 The concentration of V and Cr after 2 h electro-catalytic reaction in alkaline media |
利用UV-Vis光谱和ESR波谱等手段, 验证了在氧气存在的情况下, 碱性介质电化学催化体系中通过两电子氧还原反应生成了H
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|