
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函的甲醇和乙醇荧光特性研究
[朱从海1, 3  , 陈国庆
, 陈国庆1, 3, * , 朱纯1, 2, 3 , 赵金辰1, 3 , 刘怀博1, 3 , 张笑河1, 3 , 宋鑫澍1, 3 ]
, 陈国庆, 朱纯|
|


作者简介: 朱从海, 1993年生, 江南大学理学院硕士研究生 e-mail: zchky2015@126.com
使用英国爱丁堡FLS920P荧光光谱仪, 对甲醇、 乙醇的紫外吸收光谱和荧光光谱进行实验检测, 得到二者的光谱特征参数。 分别采用密度泛函理论(DFT)和单激发组态相互作用(CIS)对二者分子的基态和激发态结构进行优化, 并比较两种分子在不同能态下的差异。 应用含时密度泛函理论(TD-DFT)的不同泛函结合极化连续介质模型(PCM)在6-31++G(d, p)水平上分别计算了二者的紫外吸收光谱和荧光光谱, 与实验结果吻合。 分析了甲醇、 乙醇荧光产生机理, 同时分析了不同泛函对于计算光谱的影响。 结果表明: 甲醇、 乙醇在紫外波段有微弱吸收, 在紫外激发下能产生拉曼峰和微弱的荧光峰。 甲醇、 乙醇的吸收光谱是由里德堡激发产生, 产生荧光的轨道跃迁为σ*→π*。 泛函OLYP能够较好地重现实验吸收能, 而泛函MPWK能够较好地重现实验发射能, 并且发现不同的纯泛函计算跃迁能也具有差异性。 结果可为醇类分子的分子特性研究提供参考。
, CHEN Guo-qing, ZHU ChunThe absorption and the emission spectra of methanol and ethanol, scanned by the Edinburgh FLS920P steady-instantaneous fluorescence spectrometer, are studied on this paper. Aiming at comparison on the molecular structuresof methanol and ethanol under different states, we employ the density functional theory (DFT) and the single-excitation configuration interaction (CIS) to optimizemolecular structures under the ground and excited state. The absorption and emission spectra of methanol and ethanol on the base of 6-31++G (d, p) are estimated based on the time-dependent density functional theory (TD-DFT) with the polarized continuous model (PCM), which are in agreements with the experimental results. Furthermore, we analyze the fluorescence mechanism of methanol and ethanol, and investigate the effect caused by different exchange correlation functions on the calculated spectra. The results indicate that methanol and ethanol have weak absorption in ultraviolet regionand produce Raman band and weak fluorescence peaks through UV excitation. Meanwhile, the absorption spectra of methanol and ethanol are produced by Rydberg excitation, of which the orbit jumps from σ* to π*. Our results show that the OLYP function can reproduce the experimental absorption spectrum well and the MPWK function can predict the emission energy well. There exist differences on calculations of transition energy varying from different pure functions. Our results can provide a reliable tool to study alcohols’ molecular properties.
引 言
甲醇、 乙醇作为常用有机溶剂, 被广泛应用于光谱分析、 化学实验等。 前人已对甲醇、 乙醇分子结构做了大量的研究, 如Chris J Benmore利用同位素的方法研究了乙醇液体中的团簇结构以及分子动力学[1]。 Thorsten Schnabel等用1H核磁共振光谱结合分子模拟的方法研究了甲醇液体中的氢键[2]。 吴晓静等研究了几种盐在甲醇溶液中的荧光光谱, 并用理论计算得到盐/甲醇溶液中可能存在的簇合物[3]。 还有一些学者利用不同实验方法[4, 5, 6]结合理论计算, 得到了较多关于团簇结构和动力学特征的信息。 使用荧光分析法对甲醇, 乙醇的光谱性质以及结构的研究, 也有学者报道。 如本课题组对甲醇乙醇的光谱进行实验检测和初步分析[7, 8]。 刘莹等对于乙醇荧光峰位以及利用高斯分解荧光峰的方法对乙醇的团簇结构做了研究[9]。
目前甲醇乙醇的团簇结构未有准确的描述, 因此提出一种利用量子化学计算结合光谱实验研究甲醇、 乙醇构型的方法。 本文应用荧光光谱仪, 测出了甲醇、 乙醇液体荧光光谱, 并提取其特征参数; 其次采用量子化学计算的方法结合荧光发射光谱和紫外吸收光谱研究了甲醇、 乙醇二聚体结构, 并且研究了二者吸收谱的电子跃迁, 以及荧光发射机理。 最后, 利用不同的泛函计算二者的紫外吸收光谱和荧光光谱, 分析了不同泛函对于紫外吸收光谱和荧光光谱的影响, 为以后计算醇类物质提供了借鉴。
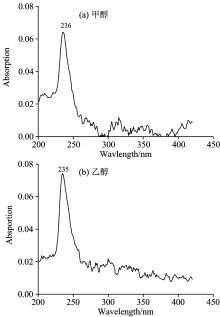
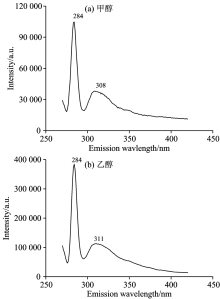
实验仪器为英国爱丁堡FLS920P稳态-瞬态荧光光谱仪。 实验样品为Sigma公司生产的甲醇和乙醇液体(纯度均大于99.9%)。 设定激发狭缝宽度为5 nm, 扫描波长范围为200~420 nm, 测量甲醇和乙醇的紫外吸收光谱, 结果如图1及表1所示。 设定激发和发射狭缝宽度为5 nm, 激发波长为260 nm, 扫描波长范围为270~420 nm, 测量两种物质的荧光发射光谱, 结果如图2及表1所示。
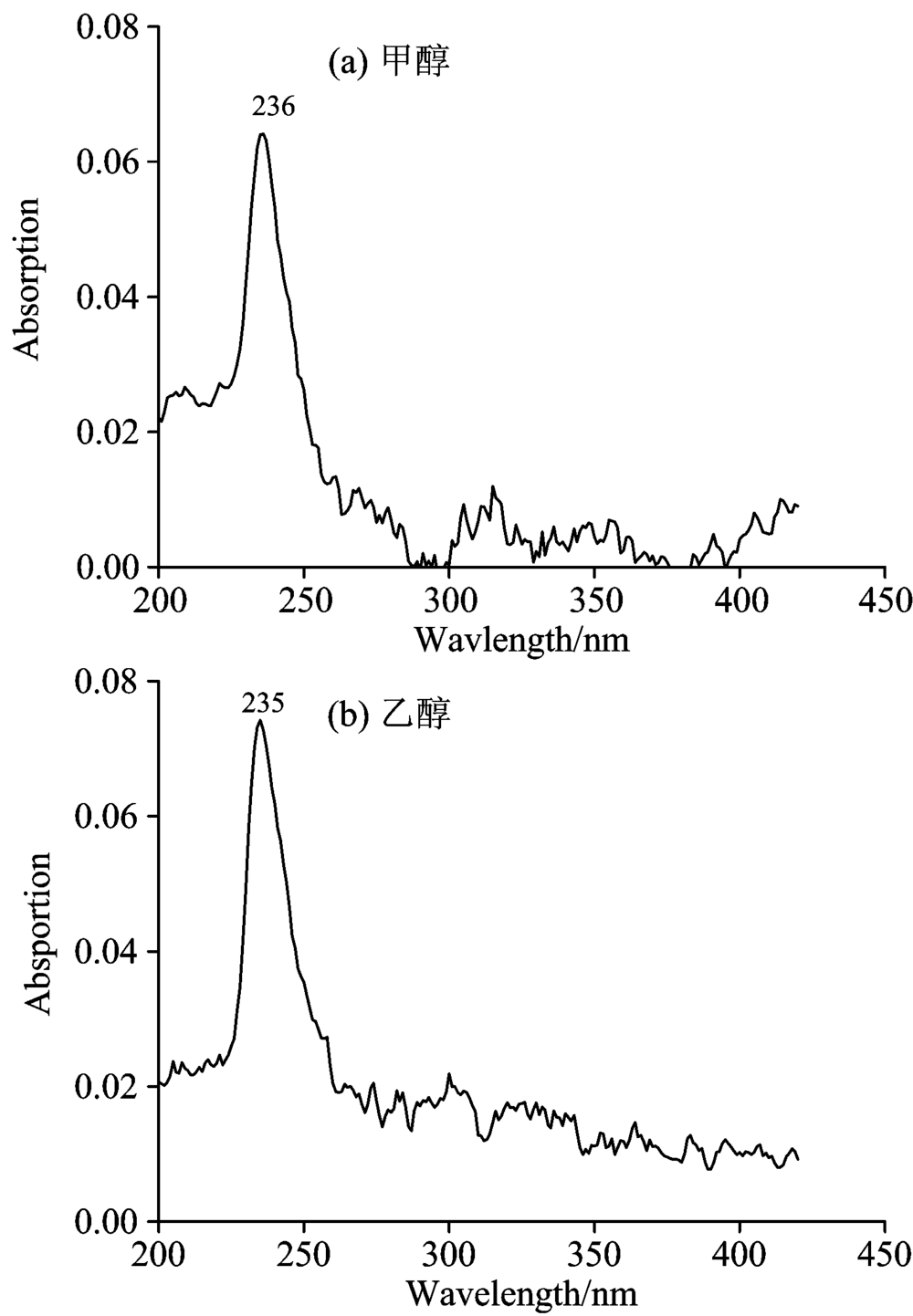 | 图1 紫外吸收光谱 (a): 甲醇; (b): 乙醇Fig.1 Absorption spectrum (a): Methanol; (b): Ethanol |
| 表1 甲醇乙醇吸收光谱和荧光光谱特征参数 Table 1 Characteristic parameters of absorption and flurescence spectra of methanol and ethanol |
实验结果显示, 甲醇、 乙醇吸收光谱的中心波长为236和235 nm, 如图1所示。 甲醇荧光峰值波长为308 nm。 284 nm处的峰经计算为甲醇碳氢键伸缩振动所产生的拉曼峰, 如图2(a)所示。 乙醇荧光峰值波长为311和284 nm处的峰经计算为乙醇碳氢键伸缩振动所产生的拉曼峰, 如图2(b)所示。
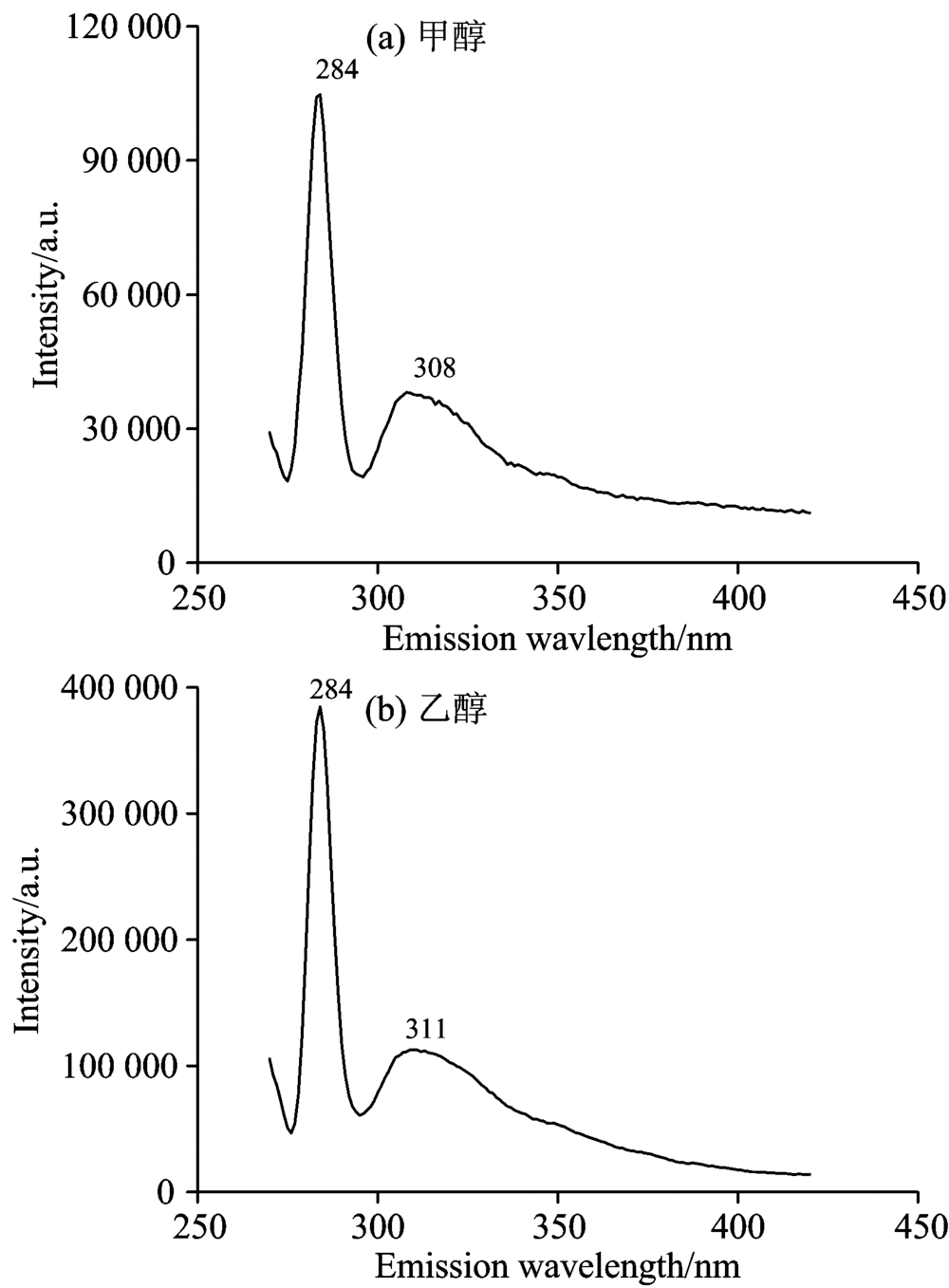 | 图2 荧光发射光谱(λ Ex=260 nm) (a): 甲醇; (b): 乙醇Fig.2 Fluorescence emission spectrum (λ Ex=260 nm) (a): Methanol; (b): Ethanol |
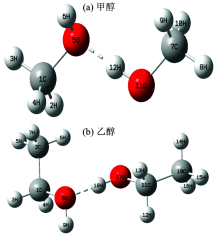
物质的光谱与其分子结构有着密不可分的关系。 通过模拟分子结构并计算其能量, 能够得到电子光谱。 考虑氢键的影响, 初始输入构型采用甲醇、 乙醇的二聚体形式如图3所示。 采用密度泛函理论(DFT)[10]中的MPWK[11], PBEPBE[12], B3LYP[13]和OLYP[14]四种泛函, 在6-31++G(d, p)基组水平上对分子的基态几何结构进行优化; 运用CIS[15]方法, 在相同基组水平上对激发态的几何结构进行优化。 通过频率分析, 发现基态与激发态优化结构无虚频, 并确认其优化结构都处于势能面的极小点。 采用含时密度泛函理论(TD-DFT)[16]和极化连续介质模型(PCM)[17], 在6-31++G(d, p)基组水平上分别计算了分子的紫外吸收光谱和荧光光谱, 并分析不同泛函对于吸收光谱和荧光光谱的影响。 甲醇、 乙醇吸收能和发射能及振子强度的理论值和实验值如表2所示。 全部计算均采用Gaussian 09[18]程序包完成。
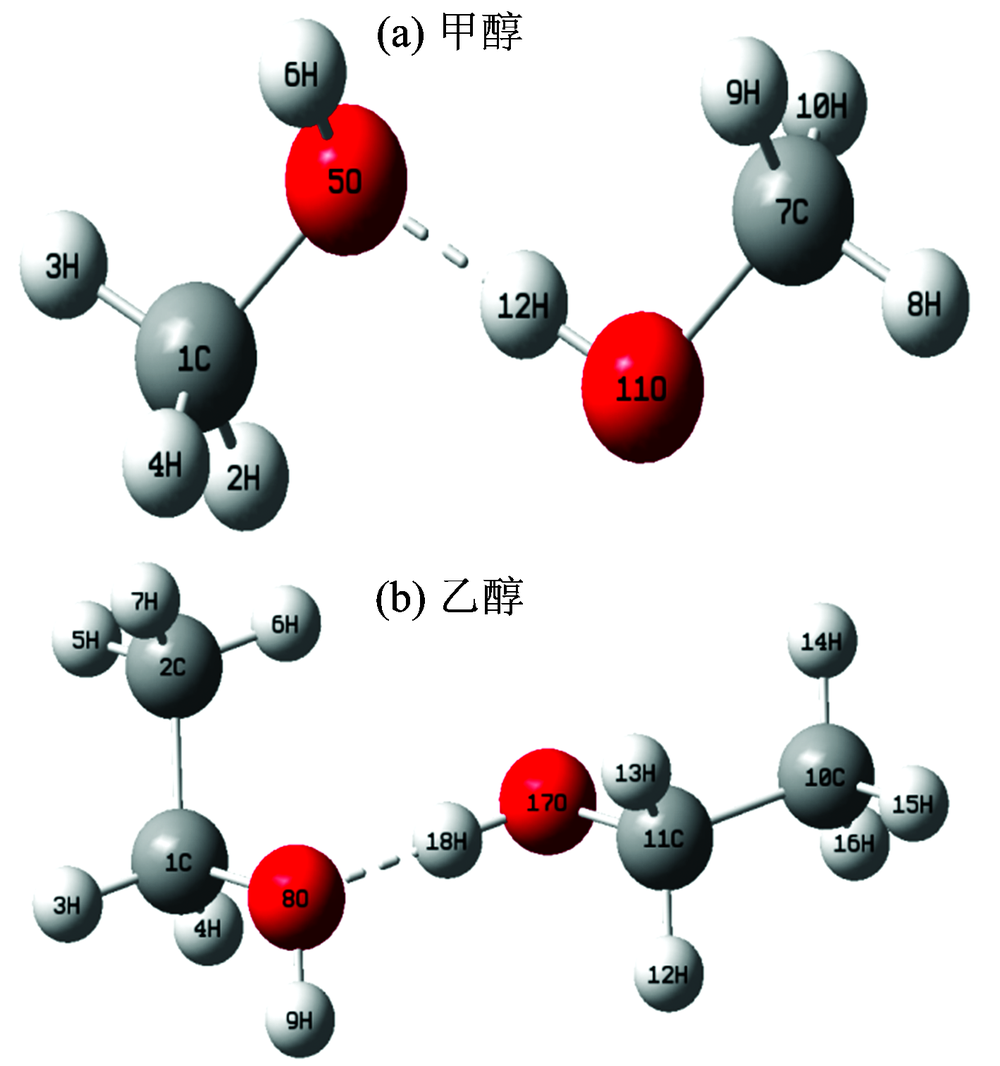 | 图3 二聚体结构 (a): 甲醇; (b): 乙醇Fig.3 Geometries (a): Methanol; (b): Ethanol |
| 表2 甲醇、 乙醇的吸收能和发射能(E)及振子强度(f)的理论值和实验值 Table 2 The absorption and emission energies (E in eV) and oscillator strengths (f in a.u.) of theoretical calculation and experimental observation |
由表2得出, 泛函OLYP和MPWK计算得到紫外吸收光谱和荧光光谱, 与实验结果最为吻合, 说明其优化的基态和激发态构型最合理。 甲醇、 乙醇分子的基态与激发态优化结构的部分参数见表3。 基态下甲醇分子的C1— H2, C1— O5和O5— H6键长分别为0.109 6, 0.143 3和0.096 7 nm, 符合正常的单键键长。 O5— O11的键长为0.301 8 nm, O5— H12— O11=174.0° , 满足氢键的形成条件。 二面角H6— O5— C1— H2=179.7° , H12— O11— C7— H8=179.7° , 说明甲醇二聚体中两个甲醇分子都有良好的共面性。 乙醇的相关键长, 键角和二面角也是类似的情况。 甲醇中的助色基团羟基本身不能吸收200 nm的光, 但它与生色团相连时, 会使生色团的吸收峰红移。 然而乙醇的最大吸收波长要比甲醇的短些, 可能是因为乙醇在碳原子的一端引入了甲基, 使得乙醇的最大吸收波长蓝移。
| 表3 甲醇、 乙醇的部分基态和激发态结构参数 Tabel 3 Part parameters of methanol and anthol molecular structure on the ground state and excited state |
激发态下甲醇分子二面角H6— O5— H12— O11=50.6° /179.9° (基态/激发态), 说明的平面程度远大于基态。 在氢键的作用下, 使两个甲醇分子的主要结构几乎形成一个平面, 这有利于荧光的产生。 相比之下, 乙醇分子在激发态下的平面性要优于基态, 但从二面角H9— O8— H18— O17=36.32° /146.7° (基态/激发态)来看, 其平面性不如甲醇。
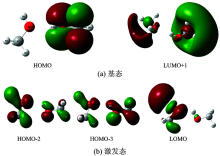
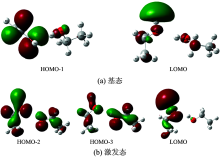
甲醇、 乙醇在OLYP/6-31G(d, p)和CIS/6-31G(d, p)水平下得到的基态和激发态的前线分子轨道如图4和图5所示。 甲醇基态的HOMO主要包括由C7的2p轨道和O11的2p轨道以肩并肩的方式形成反键轨道π * ; LUMO+1主要包括C1的
 | 图4 甲醇前线分子轨道Fig.4 The frontier molecular orbits of S0 states (a) and S1 states (b) for methanol |
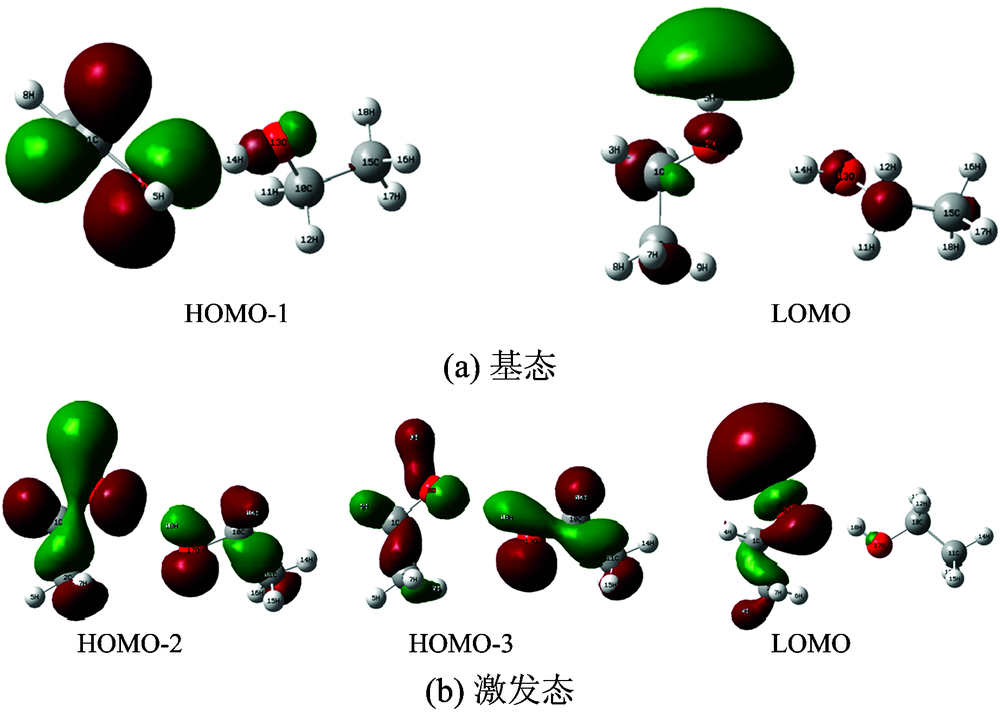 | 图5 乙醇前线分子轨道Fig.5 The frontier molecular orbits of S0 states (a) and S1 states (b) for ethanol |
与甲醇相似, 乙醇基态的HOMO-1主要包括由C1的2p轨道和O4的2p轨道以肩并肩的方式形成反键轨道π * ; LUMO主要是由H5的
不同泛函计算出的吸收能、 发射能理论值与实验值的误差如图6所示。 对于甲醇、 乙醇的紫外吸收光谱, 作为比较经典的B3LYP泛函, 理论计算的结果十分不理想, 绝对误差达到了1.60/1.41 eV, 可能因为B3LYP不适合用于计算弱相互作用。 众所周知, 激发能的TD-DFT计算结果主要取决于泛函的选择[20], 因此尝试不同泛函进行计算, 发现纯泛函对于甲醇、 乙醇的吸收能计算最接近实验值。 于是本文采用纯泛函PBEPBE和OLYP来计算吸收能, 发现即使是纯泛函, 计算的结果也具有差异性。 其中用OLYP泛函计算的结果与实验最为吻合。
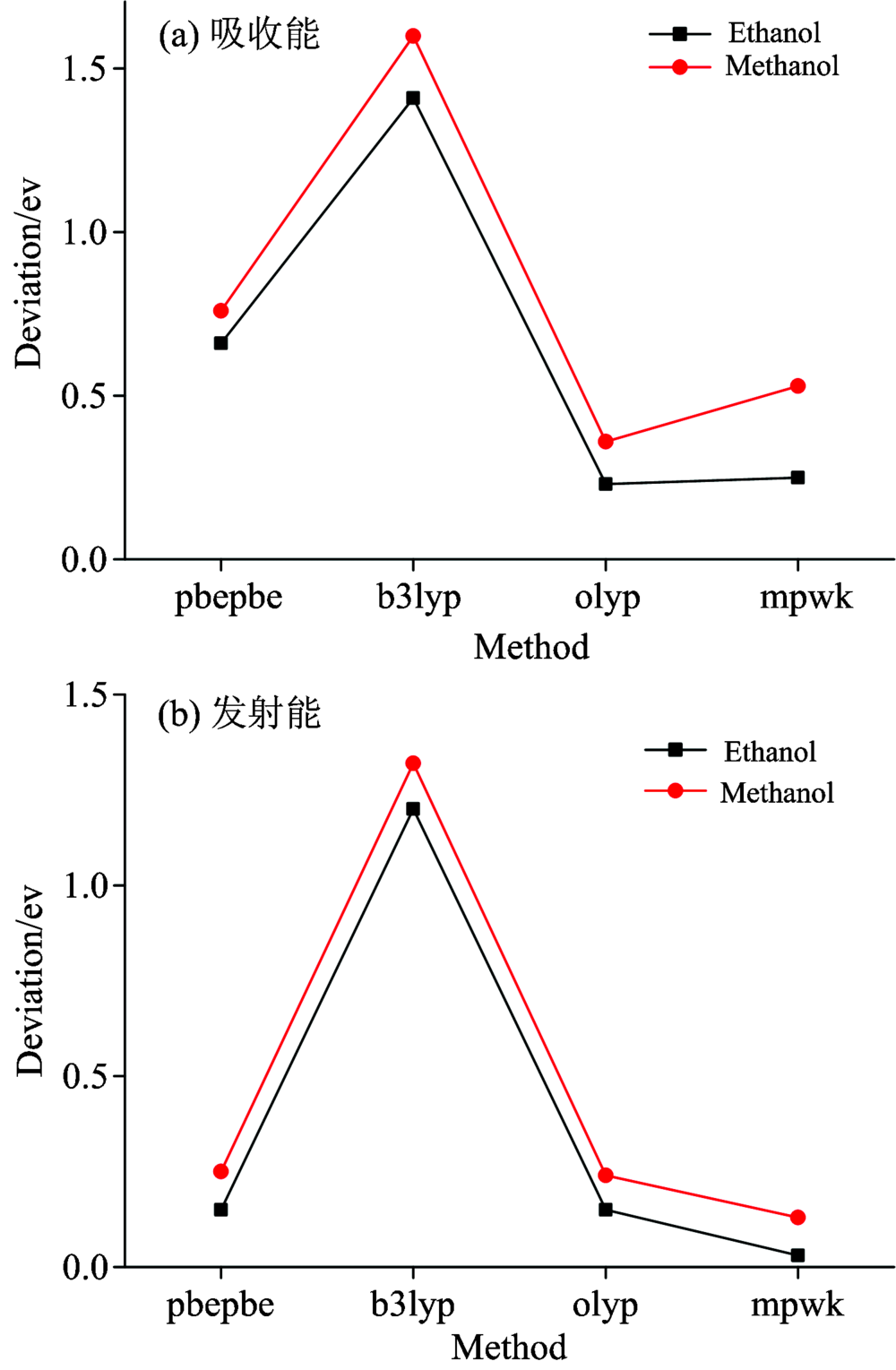 | 图6 不同泛函的计算值与实验值的对比3.2 甲醇乙醇光谱产生机理Fig.6 (a) Deviation of theoretical absorption energies to the experimental data; (b) Deviation of theoretical emission energies to the experimental data |
对于荧光发射光谱, 各种纯泛函计算的发射能相比于其计算的吸收能的结果更精确, 其中泛函MPWK计算发射能的误差仅为0.13/0.03 eV。 这说明溶剂环境对于分子激发态的构型以及电子云的分布也有很大影响, 从而影响计算结果的准确性。
根据前线分子轨道分析, 甲醇吸收光谱的电子跃迁轨道能级为HOMO→ LUMO+1, 对应的跃迁为π * →
分析表明甲醇、 乙醇吸收光谱的电子跃迁都是由C的2p轨道和O的2p轨道形成的反键轨道π * , 跃迁到弥散的里德堡轨道, 这需要非常高的能量, 因此两者的吸收波长都在紫外区域。 激发态的分子前线轨道表明, 两者荧光光谱的电子跃迁都是H的2s轨道和O的2p轨道形成的反键轨道σ * , 跃迁到C的2p轨道和O的2p轨道形成的反键轨道π * 。 由此说明两者的发射能差异不大, 符合实验结果。
实验测出甲醇、 乙醇的吸收波长分别为236和235 nm。 两者差异很小, 略微差异的原因可能是乙醇分子比甲醇多出一个甲基, 因此基态下分子平面性差于甲醇, 导致吸收光子难度增加, 需要更多的能量。 从荧光光谱来看, 激发态下甲醇的二面角H6— O5— H12— O11=179.9° , H6— O5— H12— O11=179.9° , 乙醇的二面角H9— O8— H18— O17=146.7° , C1— O8— H18— O17=13.4° , 其平面性不如甲醇。 当分子从激发态跃迁回到基态时, 平面性较差的乙醇分子经历了更多的振动和转动, 损耗了更多的能量, 因此产生荧光光子的能量减少, 即比甲醇分子的荧光波长稍长一些。
(1)甲醇、 乙醇能够吸收210~270 nm波段的紫外光, 并发射出微弱荧光。 其中甲醇、 乙醇结构并无生色基团, 能够发射290~340 nm波段的荧光, 主要因为在氢键的作用下, 分子的刚性得到了加强, 使其发出弱荧光。
(2)用泛函OLYP计算得到的吸收峰和用泛函MPWK计算得到的荧光峰值波长, 与实验值吻合, 说明其优化甲醇、 乙醇的基态和激发态结构合理。 计算得到键长、 键角及二面角等信息, 为确定甲醇、 乙醇团簇结构提供了参考。
(3)甲醇吸收光谱是由里德堡激发产生, 对应的跃迁为π * →
本文应用不同的密度泛函计算了甲醇、 乙醇的紫外吸收光谱和荧光光谱, 分析了几种方法的差异, 并与实验对比, 筛选出最合适的计算方法。 本工作对甲醇、 乙醇分子团簇结构和动力学特征的研究具有参考价值, 也为醇类物质的量子化学计算提供了借鉴。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|