{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CO探针原位红外光谱研究Pt/Sn双金属重整催化剂
[郝花花1, 2  , 袁蕙
, 袁蕙1, 2 , 徐广通1, 2, * , 向彦娟1, 2 , 马爱增1, 2 ]
, 袁蕙, 向彦娟|
|


作者简介: 郝花花, 女, 1989年生, 石油化工科学研究院硕士研究生 e-mail: huanyingzhuxin@163.com
催化重整是生产芳烃原料和高辛烷值清洁汽油调和组分的重要工艺。 以目前应用广泛的铂锡工业重整催化剂金属含量为参比, 用工业剂制备方法合成了Pt含量为0.6%的一系列铂锡重整催化剂, 建立CO探针原位红外的表征方法, 并对其进行系统表征, 首次获得了1%以下低含量助剂Sn的CO探针红外谱图。 研究结果表明, 系列剂的金属Pt的CO吸附特征峰主要以线式吸附状态存在。 0.6%纯Pt剂上1 826 cm-1处CO桥式吸附特征峰, 因添加助剂Sn后, 强度下降, 而CO线式吸附特征峰的强度则增加, 说明Sn的加入使得Pt的分散度增加。 变温CO探针吸附原位红外研究表明, 对负载质量分数0.3%的纯Sn催化剂, 当脱附温度升高至120 ℃时, 吸附在Sn上的CO特征峰会完全消失。 对负载质量分数0.6%的纯Pt催化剂, 当脱附温度升高至300℃时, 吸附在Pt中心上的CO特征峰会完全消失。 当Pt-Sn双金属负载质量分数Pt为0.6%、 Sn为0.3%时, CO的脱附温度明显提高达350 ℃。 与纯Pt剂相比, 随着Sn助剂的加入, 使得CO的脱附温度稍有提升, Pt-Sn催化剂Pt的CO特征峰向高波数方向移动, 说明Sn的加入一定程度上减弱了活性金属Pt中心上的电荷密度。 因而, CO探针原位红外是表征低金属铂锡工业重整催化剂的有效手段, 为阐明多金属重整催化剂的助剂作用和研究反应机理提供重要信息。
Catalytic reforming, which produces raw material of aromatic hydrocarbon and high-octane clean-gasolineblending component, is a principal technology. According to the content of platinum and tin loading on industrial reforming catalyst, platinum loading was kept constant at 0.6 Wt% and the amount of tin was varied from 0 to 1 Wt% in the catalysts. A series of Pt-Sn/Al2O3 reforming catalysts were characterized with in situ FTIR of adsorbed CO method which is established in this study. And the adsorbed CO infrared peaks were obtained at room temperature on Sn/Al2O3 catalysts that the mount of tin was under 1 Wt% in the first time. At room temperature the CO species of Pt sites adsorbed on the surface of Pt-Sn/Al2O3 reforming catalysts mainly on linearly-bonded CO species. It was found that there was a general trend that the amount of bridge-bonded CO species was decreasing and the amount of linearly-bonded CO species was increasing, with increasing amounts of tin in the catalyst. It was stated that the dispersity of platinum atoms was increasing. The temperature increasing desorption process was used to show that the CO band of Pt sites disappeared completely at 300 ℃ on 0.6%Pt/Al2O3, and the CO band of Sn sites disappeared completely at 120 ℃ on 0.3%Sn/Al2O3. The CO band of Pt sites and Sn sites disappeared completely at 350 ℃ on 0.6%Pt-0.3%Sn/Al2O3. Compared reforming catalysts Pt-Sn/Al2O3 with Pt/Al2O3, the linearly adsorbed CO band of Pt sites shift to high frequencies. This suggests that the electronic density of Pt increased due to the loading of Sn. Therefore, in situ FTIR is an effective tool for characterization of low metal reforming catalysts, which provides important information of interactions between tin and platinum.
石油炼制中的重整工艺是通过催化剂的作用将石脑油原料中的饱和烃转变为芳烃的技术, 是生产芳烃和高辛烷值清洁汽油调和组分的重要工艺。 最早应用的单金属型铂重整催化剂虽具有良好的芳构化性能, 但存在寿命短、 Pt易烧结、 结焦、 分散性差等问题[1, 2]。 20世纪八九十年代, 我国陆续开发了Pt-Re双金属半再生重整催化剂和Pt-Sn双金属连续重整催化剂以满足重整技术发展的需要[3]。 由于Pt-Sn体系能够抑制催化剂表面发生的加氢裂解、 低聚、 结焦等反应, 相比单Pt和Pt-Re体系, 有更好的芳烃选择性和较长的运行周期, 近年来连续重整技术得到了更快的发展[4, 5, 6], 但对Pt/Sn催化体系中助剂的作用机理的认识一直不清晰。 一般归结为空间效应和电子效应的影响。 前者认为锡的加入起到了稀释铂原子的作用, 减少了相邻铂原子的数量, 进而减少了铂原子形成产生焦炭前驱体的活性中心的可能[1], 后者则认为助剂Sn的存在可以修饰铂原子的电子环境, 电子从锡原子移向铂原子, 使得Pt-C键变弱, 进而增强了催化剂抑制焦炭生成的能力[6]。
由于CO良好的配位特性, 使其成为催化剂表征中最常用的探针分子, 根据CO与催化剂上金属活性中心的吸附行为和活性中心周围电子环境的不同导致的红外光谱特征的变化(包括谱带位置、 数目、 强度等), 可以区别不同类型的活性中心, 据此可推测催化剂活性中心的类型和催化反应机理。 Geomar等[7]曾用CO探针原位红外光谱研究了焦化处理的铂锡催化剂失活机理。 Imen等[8]通过Pd-Sn/Al2O3的CO红外光谱计算得出CO吸附热, 进而研究Pd-Sn间的作用机理。 Krishnan等[9]采用与本课题相同的催化剂体系, 通过CO探针红外光谱研究铂含量为1.0%时, 分别加入助剂锡含量0~5%后对活性中心铂的影响, 通过原位结果表明, Sn在加入1%后, 在Pt-Sn/Al2O3体系中几何效应起主导作用, 电子效应影响很弱。 考虑Krishnan等的研究结论是基于催化剂中的Pt和Sn含量在1.0%及其以上得出的, 而实际工业Pt-Sn/Al2O3重整催化剂中的Pt和Sn含量均低于1 Wt%, 这种研究体系与实际体系间组成含量的差异可能使研究结论出现偏差。 为此, 本工作以工业重整催化剂的含量范围为参照, 合成了0.6 Wt%含量Pt的Pt-Sn/Al2O3催化剂, 建立了CO探针原位红外表征方法, 表征了Pt-Sn/Al2O3催化剂, 结合表征的0.3%Sn/Al2O3CO探针原位红外的图谱特征, 考察了Pt-Sn/Al2O3的CO图谱特征变化, 以获得微量助剂存在对催化剂Pt活性中心的影响。
以Al2O3小球为载体, 采用与工业剂一致的浸渍法制备不同Pt和Sn质量分数的催化剂样品, X射线荧光光谱分析(XRF)结果详见表1。
| 表1 催化剂样品组成 Table1 The composition of reforming catalysts |
1.2.1 原位红外光谱系统
傅里叶变换红外仪为美国Thermo Fisher公司生产的NICOLET 6700, 光谱分辨率4 cm-1, 扫描次数64。
真空系统: 由两级机械泵加一级分子泵组成, 真空度可达1.0× 10-4 Pa, 石科院自行设计组装。
原位样品池: 采用石科院自行设计加工石英红外样品池, 选用氟化钙窗口, 样品池耐受温度范围在-190~500 ℃之间。
1.2.2 CO探针原位红外光谱的表征
压制直径14 mm质量12 mg范围内的催化剂样品放入原位样品池中, 以40 mL· min-1的氢气进入样品池中恒温400 ℃还原约2 h(升温速率4 ℃· min-1)。 再用250 ℃真空净化样品(真空度1.6× 10-4 Pa)为测量样品背景; 然后在室温下吸附净化后的CO, 平衡30 min后进行低真空脱附, 再次测量红外光谱。 两次测量的差谱即为CO吸附的红外特征光谱。 研究中所有谱图均将为以10 mg为标准进行质量校正。
为了与Pt-Sn体系催化剂进行对比并提取探针分子的光谱变化特征, 首先对Pt和Sn单金属的参比剂进行表征。
2.1.1 单金属Pt的CO探针红外光谱特征
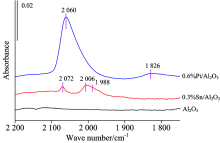
图1分别给出了催化剂Pt质量分数为0.6 Wt%不含Sn时、 Sn质量分数为0.3 Wt%不含Pt时和氧化铝载体的 CO探针红外光特征谱图。 当Pt达0.6 Wt%时, 则在1 826和2 060 cm-1出现两个特征峰, 说明当Pt在该含量时CO与Pt的结合除线式配位形态2 060 cm-1外, 部分已呈现了桥式配位吸附状态1 826 cm-1 [10], 说明此时部分Pt中心已出现聚集现象。
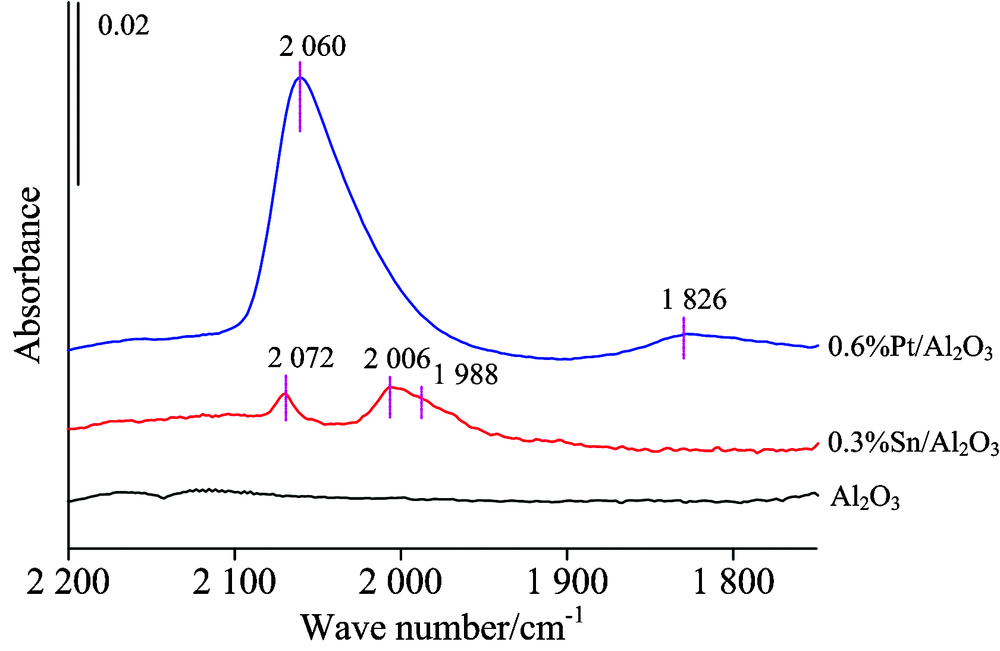 | 图1 0.6%Pt/Al2O3, 0.3%Sn/Al2O3和Al2O3的 CO探针原位红外特征图谱Fig.1 In situ FTIR spectra of CO adsorption for 0.6%Pt/Al2O3, 0.3%Sn/Al2O3 and alumina |
2.1.2 单金属Sn的CO探针红外光谱特征
图1结果显示Al2O3载体在常温下并没有出现CO吸附峰, 当载体表面负载锡含量为0.3 Wt%时, CO在常温下吸附在2 072和2 006 cm-1处直观有两个吸附峰, 说明这两个吸附峰是吸附在金属Sn的CO特征峰, 2 006 cm-1吸附峰上1 988 cm-1处有个肩峰。 可以看出, 含Sn量为0.3%的Sn/Al2O3在2 006 cm-1的峰高在0.004左右, 虽然与0.6%纯Pt剂Pt中心上的CO吸附强度相比弱, 但在金属Sn中心的吸附峰不可忽略。 但以往文献对Sn的CO吸附情况鲜少提及, 没有明确的归属[8, 9, 11]。
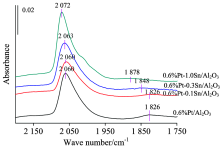
固定Pt的含量0.6%, 在不同含量水平上改变负载Sn含量, 再对比纯Pt剂和纯Sn剂的CO吸附特征峰, 观察Sn含量的变化对催化剂红外特征峰的影响, 结果见图2。 由图可以看出, 当催化剂添加Sn后, 所测量的是CO在双金属催化剂Pt中心和Sn中心吸附的叠加结果。 即在Pt中心上的线式吸附态特征与Sn中心上约2 070 cm-1特征峰的叠加, 表观上最大吸收峰略向高波数位移动。 重要的是Pt中心桥式吸附态特征的变化, 可以看出, 随Sn加入量的增加, 该特征峰的峰高、 峰面积逐渐变小, 吸附峰甚至有消失的现象, 同时峰位从1 826 cm-1不断蓝移到1 878 cm-1, 表明少量Sn的加入明显“ 稀释” 了Pt原子的团聚, 使得Pt的分散性增强, 助剂金属的加入呈现出明显的几何效应。
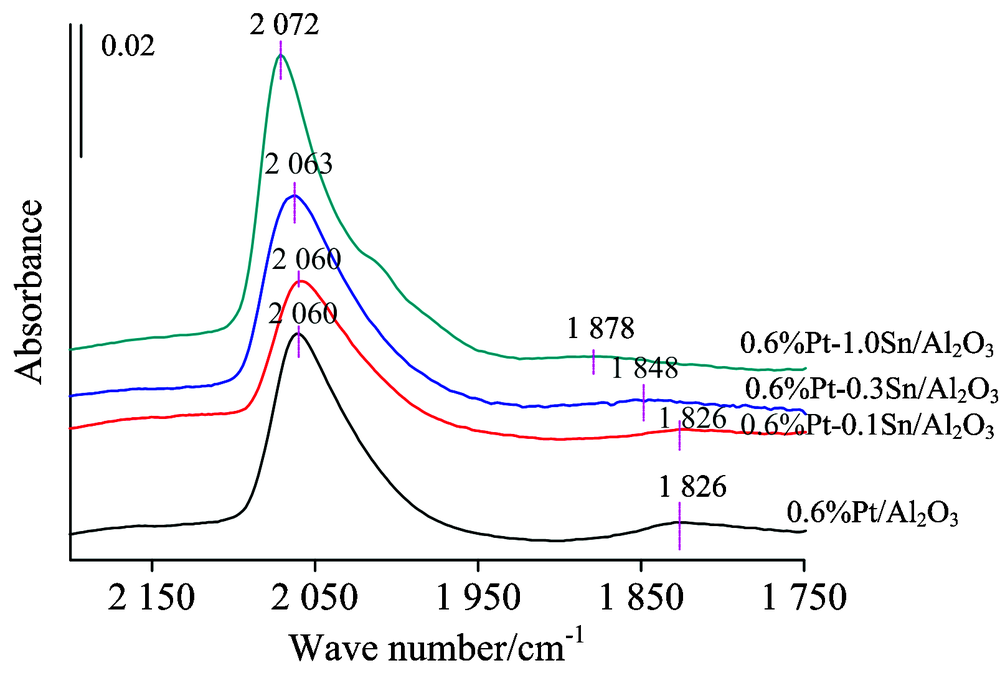 | 图2 铂含量0.6%锡含量变化的系列 催化剂的CO探针原位红外图谱Fig.2 In situ FTIR spectra of CO adsorption for series catalysts |
为研究双金属催化剂中Pt-Sn之间相互作用的电子效应, 采用变温CO探针实验比较了0.6%Pt/Al2O3, 0.6%Pt-0.3%Sn/Al2O3和0.3%Sn/Al2O3时吸附CO的红外光谱特征变化情况。 三个样品的变温脱附实验结果见图3、 图4、 图5。
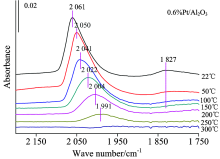
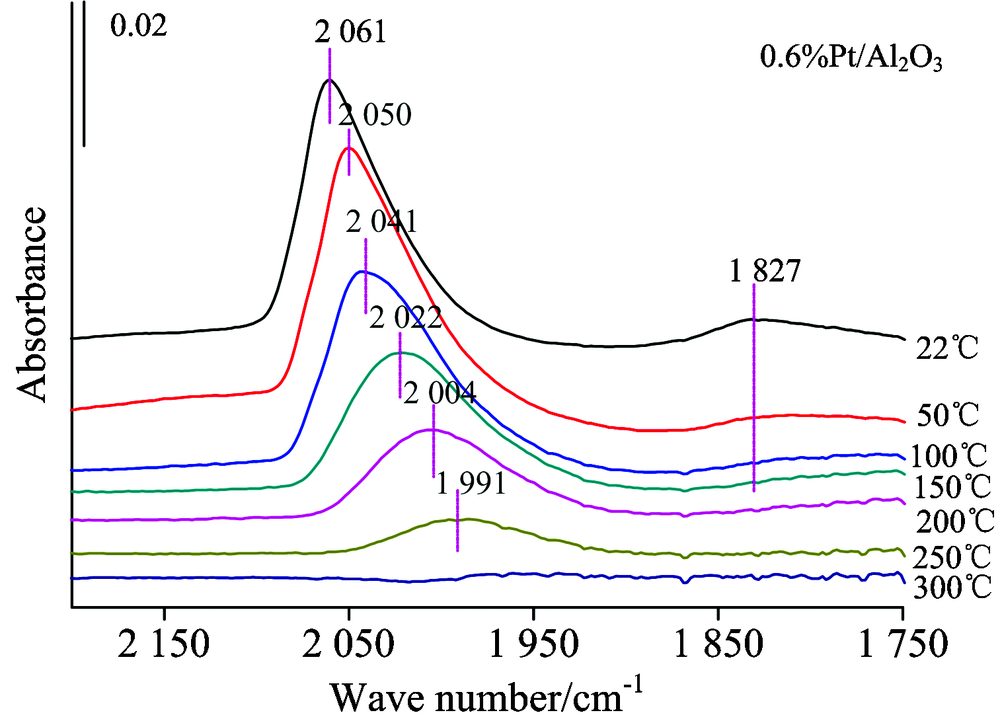 | 图3 样品0.6%Pt/Al2O3的原位红外变温实验图谱Fig.3 FTIR spectra collected during the desorption of CO from 0.6%Pt/Al2O3, with treatment in high vacuum at various temperature |
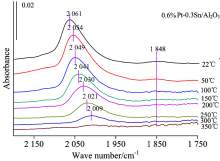
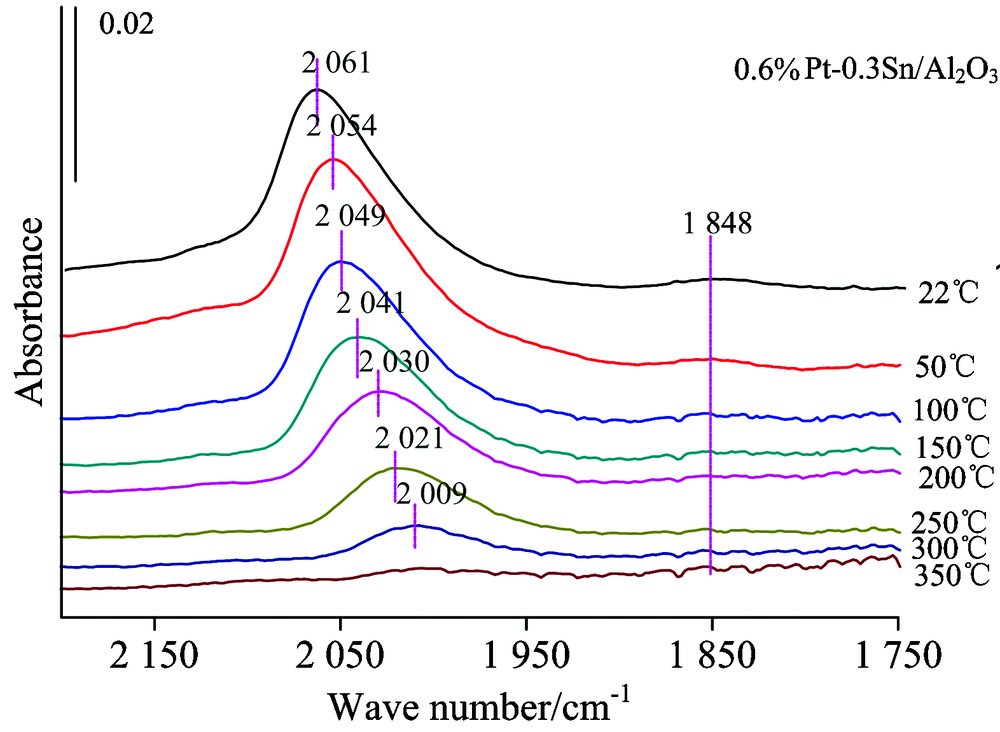 | 图4 0.6%Pt-0.3%Sn/Al2O3的原位红外变温实验图谱Fig.4 FTIR spectra collected during the desorption of CO from 0.6%Pt-0.3%Sn/Al2O3, with treatment in high vacuum at various temperature |
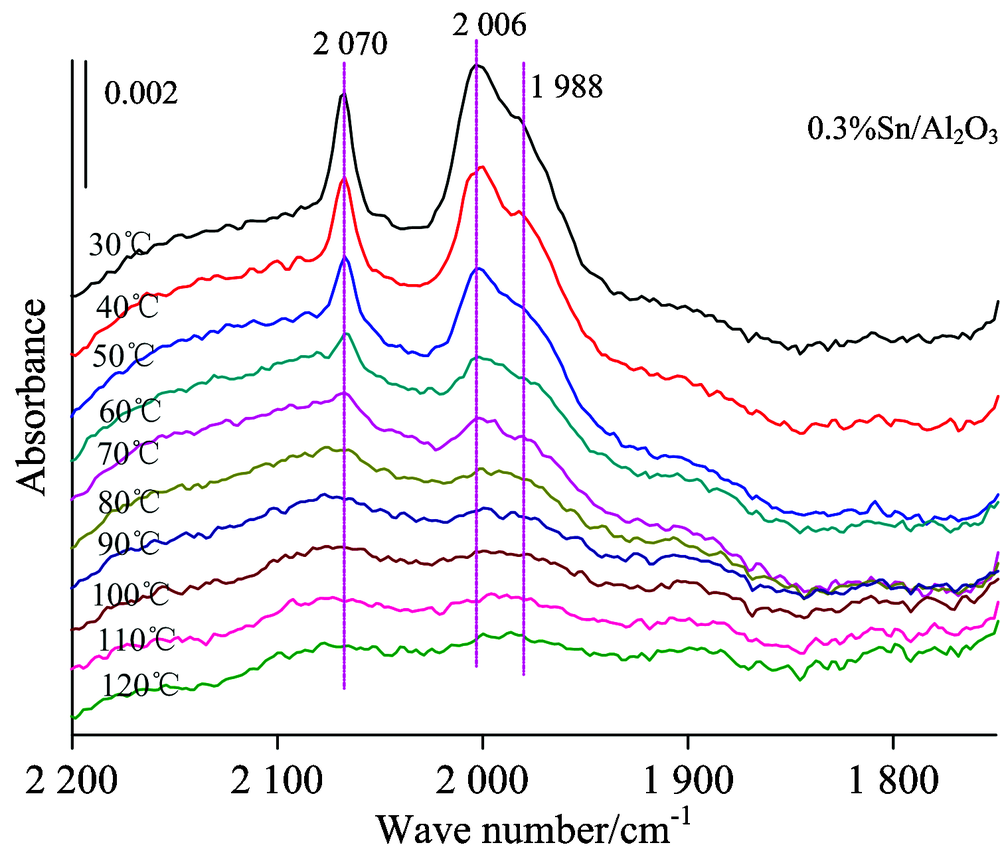 | 图5 0.3%Sn/Al2O3的原位红外变温实验图谱Fig.5 FTIR spectra collected during the desorption of CO from 0.3%Sn/Al2O3, with treatment in high vacuum at various temperature |
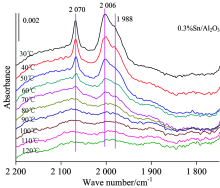
图3为仅负载Pt的样品的CO变温脱附的系列红外光谱。 可以看出随着温度的升高, 最先消失的是在Pt中心上CO的桥式吸附峰; 当升温至100 ℃时桥式吸附态已基本消失, 而在Pt中心上的CO呈线式的吸附态在300 ℃时才基本消失, 说明CO与Pt中心呈桥式吸附的强度相比线式吸附态较弱。 2 060 cm-1左右的Pt线式吸附峰在温度不断升高的过程中吸附峰高急剧降低、 峰位大幅度红移。 图4为添加Sn后催化剂吸附CO的变温脱附谱图, 此时Pt的CO线式吸附峰在350 ℃时基本消失, 其他与0.6%Pt/Al2O3规律相似, 说明相比纯Pt剂, Sn的加入使得CO与Pt中心的吸附强度有所增强。
图5则为载体上仅负载助剂Sn时吸附CO的变温脱附红外谱图, 可以看出随着温度升高, Sn的两个吸附峰都不断减小, 在120 ℃时基本脱附完全, 但与Pt中心上吸附的CO不同, Sn中心上吸附CO的峰位随着温度的上升没有明显变化。
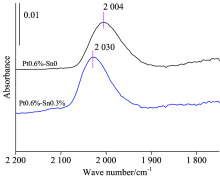
综合比较分析CO探针在三个不同金属负载体系上CO变温脱附红外光谱特征的变化情况, 可以看出当温度超过120 ℃时CO在Sn中心上基本完全脱附, 超过此温度可以获得CO仅在Pt中心上的吸附状态, 也更接近于重整反应的温度区。 图6为200 ℃时CO分别在0.6%Pt/Al2O3和0.6%Pt-0.3%Sn/Al2O3两个催化剂上的吸收特征, 可以看出Pt线式吸附峰分别在2 004和2 030 cm-1处, 负载0.3%的Sn后Pt的CO线式吸附峰位蓝移26 cm-1。 说明随Sn的加入使Pt原子上电荷密度减小, 金属中心上的d电子反馈减弱, Sn助剂表现出一定的吸电子效应。 这也解释了工业Pt/Sn双金属重整催化剂在性能上表现为随Sn的加入在芳构化性能上略有下降, 但由于Sn几何效应的存在, 抑制了Pt的聚集, 使催化剂的运行寿命得到很大的提升。
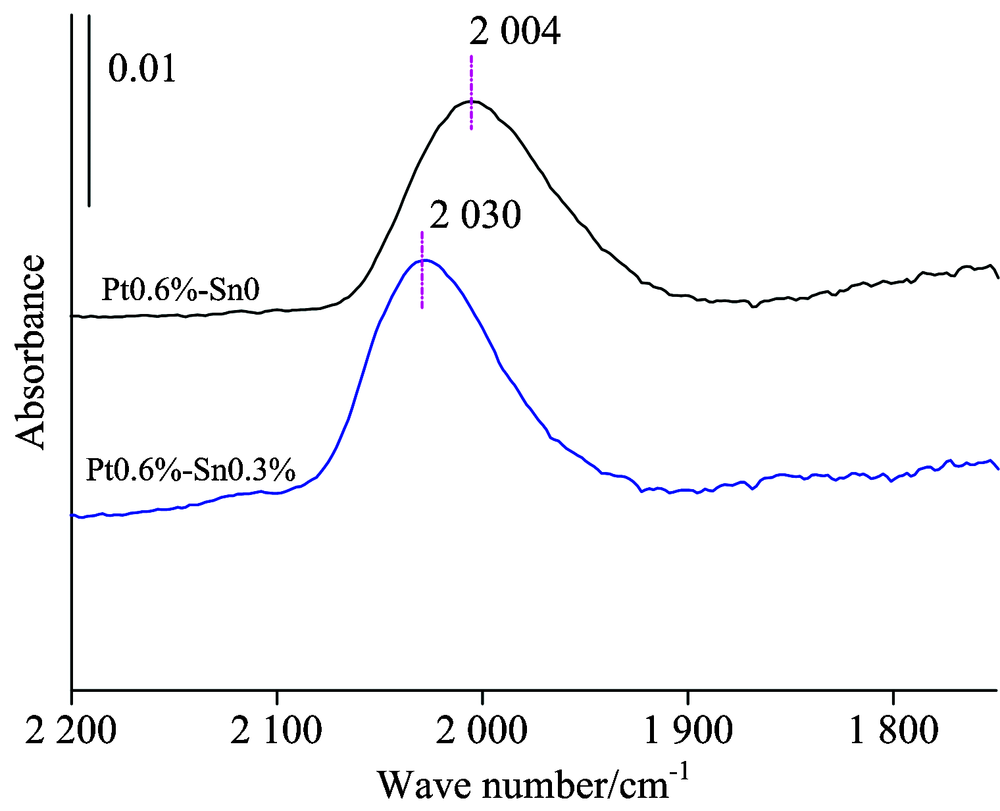 | 图6 0.6%Pt催化剂系列在200 ℃时的脱附曲线图Fig.6 FTIR spectra collected during the desorption of CO from 0.6%Pt/Al2O3, 0.6%Pt-0.3%Sn/Al2O3 reduced at 200 ℃, with treatment in high vacuum |
(1)当单Pt剂中Pt的负载含量达0.6%时, CO探针红外谱图上1 826 cm-1处出现明显的桥式吸附。 首次检测到0.3%锡含量的纯Sn剂在CO探针红外谱图上存在2 006与2 072 cm-1两个吸附峰和1 988 cm-1处的小肩峰。
(2)当催化剂中添加助剂Sn后, 呈桥式吸附的谱带强度下降, 桥式吸附峰在1.0%Sn含量时甚至趋于消失, 而呈线式吸附的谱带强度明显增强, 说明即使少量助剂Sn的加入也使活性金属Pt的分散性增加, 具有一定的几何效应。
(3)变温CO探针吸附红外谱图特征表明, 单Pt剂(0.6%)的CO脱附温度为300 ℃, 0.3%锡含量的纯Sn剂的CO脱附温度为120 ℃, 加入0.3%Sn后的Pt/Sn重整剂的CO脱附温度为350 ℃。 当温度超过120 ℃时, CO探针在助剂Sn上的吸附已经消失, 比较200 ℃下助剂Sn含量为0.3%时CO在Pt中心的吸附特征可以看出有助剂存在时CO的吸收向高频区移动, 说明Sn的存在对Pt中心有一定的吸电子效应, 导致Pt中心的电荷密度有所降低。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|