

{kind=link}
{kind=link}
{kind=link}
{kind=link}
系列二亚胺羰基Mn配合物光谱的密度泛函研究
[慈成刚*  , 臧杰超, 李明飞
, 臧杰超, 李明飞* ]
, 臧杰超, 李明飞|
|


作者简介: 慈成刚, 1982年生, 黔南民族师范学院贵州省计算催化化学重点实验室副教授 e-mail: chenggci@sgmtu.edu.cn
CO2还原始终是能源和环境领域的重要挑战。 二亚胺羰基Mn配合物价格低廉, 稳定性好, 可调变性强, 成为近年来光催化还原CO2的热门催化剂。 紫外-可见光谱和红外光谱研究有助于调控CO2光还原催化剂性能。 基于密度泛函理论(DFT)和含时密度泛函理论(TD-DFT), 对系列二亚胺羰基锰配合物[Mn(bpy)(CO)3Br], (简写为1), [Mn(phen)(CO)3Br], (简写为2), [Mn(phen-dione)(CO)3Br], (简写为3), [Mn(phen-dione)(CO)3CH3CN]+, (简写为4) (bpy=2,2'-bipyridine, phen=1,10-phenan-throline, phen-dione=phenanthroline-5, 6-dione)的紫外-可见光谱和红外光谱进行研究。 基于TD-DFT方法, 采用多种泛函, 对紫外-可见光谱进行模拟。 结果显示1和2主要有两个最大吸收峰, 分别位于371 nm (1), 408 nm (1)和361 nm (2), 414 nm (2), 其电子跃迁类型均为由金属Mn中心基团向二亚胺配体的电荷转移(MLCT)跃迁。 而3和4均具有三个吸收峰, 分别位于290 nm (3), 337 nm (3), 431 nm (3)和294 nm (4), 319 nm (4), 371 nm (4)。 其中, 除了4的294 nm吸收峰对应了二亚胺配体内部的π—π*跃迁, 3和4的其余吸收峰均为MLCT跃迁。 伴随着二亚胺配体电负性的增强, 吸收峰向可见光区移动(红移), 而因Mn中心配体电负性的增强, 导致吸收峰向紫外光区移动(蓝移)。 一旦电子从Mn中心基团转移至二亚胺配体, Mn中心基团成为缺电子中心, 有利于外界电子进入。 因Mn中心基团的轨道主要由金属Mn和配体的 σ*反键轨道组成, 有利于Mn中心基团配体Br-/CH3CN解离, 形成活性中间体。 红外光谱计算结果显示1, 2, 3和4的特征振动峰主要分为两类: 金属Mn中心的C=O键伸缩振动(1, 2, 3和4的1 920~2 020 cm-1)和二亚胺羰基的C=O键伸缩振动(3的1 690 cm-1和4的1 694 cm-1)。 伴随着二亚胺羰基配体和Mn中心配体电负性的增强, 1到4的特征峰波数略微增加。 计算的分子结构, 紫外-可见光谱和红外光谱与实验结果符合很好, 能够为二亚胺羰基锰配合物的合成和光还原CO2性能调变提供可靠的理论参考。
The reduction of CO2 is still a challenge in energy sources and the environmental field. Diamine coordinated manganese tricarbonylcatalysts containing diamine ligands, as inexpensive molecule based inorganic materials, become a potentially interesting catalyst in the photoreduction of CO2. The ultraviolet-visible (UV-Vis) and infrared (IR) spectra are beneficial to the modulation of catalysts for the photoreduction of CO2. TheUV-Vis and IR spectra of a series of diamine coordinated manganese tricarbonyl catalysts, [Mn(bpy)(CO)3Br], (1), [Mn(phen)(CO)3Br](2), [Mn(phen-dione)(CO)3Br](3), [Mn(phen-dione)(CO)3CH3CN]+(4) (bpy=2,2'-bipyridine, phen=1,10-phenan-throline, phen-dione=phenanthroline-5,6-dione) has been investigated using Density Functional Theory (DFT) and Time-Dependent Density Functional Theory (TD-DFT) calculations. The UV-Vis spectrawere obtained using several TD-DFT calculations. The simulated UV-Vis spectra of 1 and 2 all show two peaks centered at 371 nm (1), 408 nm (1) and 361 nm (2), 414 nm (2), respectively. Absorption peaks of 1 and 2 arise from a metal-to-ligand charge transfer (MLCT) transition. The simulated UV-Vis spectra of 3 and 4 show three absorption peaks centered around 290 nm (3), 337 nm (3), 431 nm (3) and 294 nm (4), 319 nm (4), 371 nm (4), respectively. Herein, except for the peak of 294 nm (4) generated by the ligand-to-ligand π—π* transition, the remain absorption peaks of 3 and 4 all arise from MLCT absorption. The increased electronegativity of diamine ligands is responsible for shifting absorption peak from the UV region to the visible region (red). The increased electronegativity of the Mn-centeredligand is responsible for shifting absorption peak from the visible region to the UV region (blue). Once an electron is transferred from the Mn-centered unit to the diamine ligands, the Mn center will become anelectron-deficient unit. Herein, the Mn-centeredunit contributes from the σ antibonding orbital between the Mn atom and ligand in the MLCT states. Thus, when the populated excited MLCT states are hit, the release of Mn-centered ligand (Br-/CH3CN) becomes favorable, and the system can form the active species. The simulated IR spectra show that two kinds of characteristic peaks are obtained, that is, (1) stretching vibration of Mn-centered C=O unit at 1 920~2020 cm-1 (1, 2, 3, and 4) and (2) stretching vibration of C=O unit of diamine ligands at 1 690 cm-1 (3) and 1 694 cm-1 (4), respectively. The increased electronegativity of diamine ligands and Mn-centered ligand strengthens the C=O bond, which increases the stretching vibration frequency of the C=O units from 1 to 4. The calculation results, including molecular structures, the UV and IR spectra show good agreement with those obtained by experiment and provide the reasonable theoretical guide for the synthesis and regulation of diamine-coordinated manganese tricarbonyl catalysts.
CO2是非常稳定且完全氧化态的碳形式, 治理和利用CO2成为能源和环境催化领域的研究热点之一[1]。 光还原CO2作为一种有效手段, 能够转换CO2为有用化学品, 降低大气中的CO2含量, 同时将太阳能存储为化学能, 实现了能源和环境领域的双赢[1]。 紫外-可见光谱和红外光谱能够探测CO2还原催化剂结构的重要信息, 成为CO2光还原机理研究的重要手段[1]。
二亚胺羰基锰配合物价格低廉, 能够通过配体结构调变其光催化性能, 在可见光下还原CO2为CO或HCOOH, 成为一种高效光催化剂[2, 3, 4]。 2014年, Takeda等报道了第一个二亚胺羰基锰光催化剂[Mn(bpy)(CO)3Br)][3]。 为了提高催化性能, 研究者们进行了大量探索[2, 3, 4]。 其中, Jean-Daniel等使用紫外-可见光谱和红外光谱等研究了[Mn(phen)(CO)3Br], [Mn(phen-dione)(CO)3L], (L=Br或CH3CN)光还原CO2[4]。 然而因电子跃迁的快速和实验技术的限制, 无法在分子层面直接探测这些催化剂的前线分子轨道, 电子跃迁特征和分子振动模式。 而这些信息对调变催化剂活性和选择性都具有重要意义。
有报道使用DFT和TDDFT方法对各类分子结构的紫外-可见光谱和红外光谱等进行了大量研究[5, 6, 7, 8, 9]。 其中, Keith等[7]和Sung等[8]对二亚胺羰基锰配合物的结构和振动频率进行了计算。 前期工作对Re取代有机多酸杂化配合物的紫外-可见光谱, 振动频率和光还原CO2机理进行了较为详细的研究[9], 为理论研究二亚胺羰基锰配合物的紫外-可见光谱和红外光谱提供了参考。 本工作使用DFT和TDDFT方法对系列二亚胺羰基锰配合物1, 2, 3, 4的结构、 前线分子轨道、 紫外-可见光谱和红外光谱进行计算。 给出电子跃迁特征和分子振动模式, 以及不同配体对紫外-可见光谱和红外光谱的影响, 为二亚胺羰基锰配合物的实验合成和光还原CO2性能调控提供参考。
所有计算均采用Gaussian 16 C.01程序包。 几何结构优化采用B3LPY杂化泛函。 对Mn和Br原子采用SDD基组, 对C, H, N和O等非金属原子采用6-311+G(d, p)基组。 对所有优化的结构都进行频率计算, 以确定极小能量结构。 使用TDDFT方法计算了50个最低电子激发态性质。 使用B3LPY, CAM-B3LYP, M06, M062X, M06HF, M06L, PBE0, LC-wPBE和wB97XD泛函模拟紫外-可见光谱。 使用GaussSum 3.0[10]绘制紫外-可见光谱, 使用GaussianVew 6.1.1绘制红外光谱。 除wb97xd以外, 对M06系列泛函使用D3色散校正, 对其他泛函使用D3BJ色散校正。 所有计算均采用严格的收敛标准(10-12 au), 并均采用IEF-PCM溶剂化模型, 乙腈为溶剂。
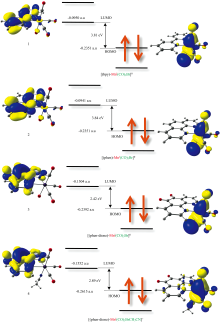
关于配合物1, 2, 3, 4光还原CO2反应曾在乙腈溶剂中进行[4], 因此使用B3LYP泛函, 在乙腈溶剂中, 对配合物1, 2, 3, 4进行全优化(图1)。 频率计算结果显示所有频率均无虚频, 即均为势能面上能量极小点。 为了更好的理解催化剂活性起源, 基于优化的结构, 计算了配合物1, 2, 3, 4的前线分子轨道(图2)。
 | 图1 计算的配合物1, 2, 3和4分子结构Fig.1 Calculated structure of 1, 2, 3, and 4 |
 | 图2 1, 2, 3和4的的前线分子轨道Fig.2 Frontier molecular orbital (FMO) of 1, 2, 3, and 4 |
计算结果显示配合物1, 2, 3, 4的最低空轨道(LUMO)均主要来自二亚胺配体中C原子和O原子2p轨道的贡献, 而最高占据轨道(HOMO)均主要由中心金属Mn的3d轨道和配体的2p轨道构成, 呈现了明显的σ * 反键轨道特征。 预测一旦受到紫外-可见光激发, 金属Mn中心电子将被激发, 形成空穴, 有利于外界电子进入其σ * 轨道, 促使配体Br-或CH3CN解离, 形成活性中间体, 推动催化反应进行。
基于B3LYP优化的基态结构, 根据Frank-Condon原理, 采用TDDFT方法, 在乙腈溶剂中, 计算了前50个激发态(图3)。 为验证结果可靠性, 使用多种泛函进行对比(表1)。 可以看出B3LYP泛函计算的最大吸收峰与实验值符合最好。 催化剂1和2的最大吸收峰均为两个, 均对应电子从金属Mn中心向二亚胺配体的电荷转移(MLCT)跃迁(图3)。 以1的371 nm吸收峰为例, 产生原因为电子从占据轨道HOMO-3向非占据轨道LUMO的跃迁。 HOMO-3轨道组成主要为Mn的3d轨道和Br的3p轨道, 而LUMO轨道主要由二亚胺羰基配体的C原子和O原子的2p轨道组成。 因此, 该跃迁类型为MLCT跃迁。 对于催化剂3和4, 因二亚胺配体中C=O基团的出现, 使最大吸收峰出现了三个。 其中, 因4的Mn中心配体为CH3CN, 其294 nm的吸收峰对应电子从占据轨道HOMO-4到非占据轨道LUMO+1的跃迁。 而HOMO-4和LUMO+1均为二亚胺配体中C原子和O原子2p轨道组成的π 轨道。 该跃迁为二亚胺配体内部的π — π * 跃迁(图3)。 3和4的其他吸收峰与1和2类似, 均为MLCT跃迁。
 | 图3 B3LYP泛函计算配合物1, 2, 3, 4的紫外光谱(单位nm)和垂直激发能(单位kcal· mol-1)Fig.3 Calculated UV spectra (in nm) and vertical excited energies (in kcal· mol-1) by means of B3LYP functional complex 1, 2, 3, 4 |
| 表1 计算的1, 2, 3和4的紫外-可见吸收峰(单位 nm) Table 1 Calculated absorption peaks (λ ) of 1, 2, 3, and 4 (in nm) |
以化合物1为例, 一旦电荷由Mn中心基团进入二亚胺配体后, 将形成具有{(bpy)e[Mn(CO)3Br]+}特征的活性结构。 而Mn中心基团为明显的Mn— Br键的σ * 反键轨道(1的HOMO-1和HOMO-3, 图3), 电子进入该σ * 反键轨道后, 将有利于配体Br-解离, 形成活性中间体, 推动反应进行。 当Mn中心配体均为Br-时, 二亚胺配体电负性的增加(1, 2到3), 使最大吸收峰向可见光区移动(1的408 nm, 2的414 nm到3的431 nm), 而Mn中心配体电负性的增强(Br-变为CH3CN), 使最大吸收峰向紫外区移动(3的431 nm到4的371 nm)。 因此, 增加二亚胺配体电负性, 降低Mn中心配体电负性, 将有利于吸收峰向可见光区移动, 提高可见光利用率。
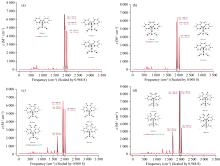
红外光谱特征峰的频率、 强度和形状能准确反映分子结构, 成为监测催化机理的有效手段。 采用B3LYP泛函, 在乙腈溶剂中, 对配合物1, 2, 3, 4结构进行全优化, 并在相同计算水平上计算了振动频率, 并采用校正因子0.968 8[11], 进行频率校正。 计算结果显示所有振动频率没有虚频, 均为势能面上的能量极小点。
计算的所有红外光谱特征峰对应的分子振动模式和相应的实验值绘制于图4(a— d)和列于表2。 计算结果和实验值符合很好, 最大误差仅为8 cm-1 (0.02 kcal mol-1)。 因配合物1, 2, 3, 4结构相似, 导致红外特征峰也十分相似, 主要归纳为两类: (1) 与金属中心Mn配位的C=O键伸缩振动(配合物1, 2, 3和4); (2) phen-dione配体的C=O伸缩振动(配合物3和4)。 伴随着二亚胺羰基配体出现C=O基团, 导致其通过诱导效应使Mn中心C=O基团中O原子电荷向C原子转移, 增强了C=O键, 提升了Mn中心的C=O振动频率, 使特征峰向高波数移动(配合物3, 4相比1, 2增加了约8~24 cm-1)。 同样, 因Mn中心配体Br-和CH3CN电负性的差异, 导致配合物4的振动峰均高于配合物3。
 | 图4 计算的红外光谱(单位cm-1)和振动模式 (a), (b), (c), (d)分别对应化合物1, 2, 3, 4Fig.4 Calculated IR spectra (in cm-1) and vibrational mods (a), (b), (c), (d) correspond to the speccies 1, 2, 3, 4 |
| 表2 1, 2, 3和4的主要红外特征峰(单位cm-1), 振动模式和摩尔吸收系数ε (单位mol-1· L· cm-1) Table 2 Calculated infrared peaks (in cm-1), major vibrational modes and molar absorption coefficient, ε (in mol-1· L· cm-1) of 1, 2, 3, and 4 |
采用DFT和TDDFT方法, 在乙腈溶剂中, 对系列二亚胺羰基锰配合物1, 2, 3, 4的结构, 前线分子轨道, 紫外-可见光谱和红外光谱进行计算。 结果显示:
(1)理论计算的几何结构, 紫外-可见光谱和红外光谱与实验值符合很好。
(2)HOMO轨道均为Mn中心基团的σ * 反键轨道, 一旦电子进入该轨道, 将有利于配体Br-或CH3CN的解离和活性中间体产生。
(3)1, 2和3紫外吸收峰的电子跃迁类型均为MLCT, 4还存在二亚胺配体内部的π — π * 跃迁。 二亚胺配体电负性增加使最大吸收峰红移, 而Mn中心配体电负性增加使最大吸收峰蓝移。
(4)配合物1, 2, 3, 4的红外特征峰主要分为两类, 即Mn中心C=O键的伸缩振动和二亚胺配体的C=O键伸缩振动。 3和4的二亚胺配体电负性的增强, 使Mn中心C=O键振动的特征峰向高波数移动, 而Mn中心配体电负性的增强, 使4的振动峰强于3。
对系列二亚胺羰基Mn配合物紫外-可见光谱和红外光谱的研究, 将为后续实验合成相关CO2光还原催化剂和调控催化性能提供帮助。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|