


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函理论的氯氰菊酯振动光谱研究
[梁小蕊1  , 丛静娴
, 丛静娴2 , 李荫1 , 刘洁1 , 金靓婕1 , 孙晓伟1 , 李晓栋3 ]
, 丛静娴|
|


作者简介: 梁小蕊, 女, 1979年生, 海军航空大学航空基础学院副教授 e-mail: xiaoruiliang12@163.com
氯氰菊酯作为一种广谱杀虫剂被广泛用于多种农产品, 由于其用量大、 降解速度慢, 导致在水果、 蔬菜及家畜等农产品中存在药物残留, 危害人体健康。 为避免人体摄入, 对农产品中的氯氰菊酯残留检测非常重要。 目前的检测方法中振动光谱技术具有无损快速的优点, 因此采用密度泛函理论方法结合振动光谱技术为氯氰菊酯的振动光谱检测和鉴定提供理论依据, 为农药残留检测应用领域提供参考。 研究中首先采用Gaussian view6.0软件构建氯氰菊酯分子的空间构型, 基于密度泛函理论DFT中的B3LYP方法, 先用3-21G基组进行初始结构粗优化, 在粗优化结构的基础上用6-311++G基组进行结构的再优化, 得到分子的最稳定构象及前线轨道分布。 然后在优化结构的基础上计算了氯氰菊酯的理论红外光谱和拉曼光谱。 理论计算结果可见氯氰菊酯分子在3 300~3 000与1 700~500 cm-1范围两个区域有明显的红外活性, 前者主要是官能团的振动, 后者则为指纹区的振动; 从计算结果还可以明显看出, 拉曼光谱中3 044和1 459 cm-1处的环丙基上亚甲基碳氢的伸缩振动、 剪式振动, 1 196 cm-1处的环丙基中次甲基的摇摆振动, 1 153 cm-1处的苯环中碳氢的面内摇摆振动等明显的振动峰, 在红外光谱中没有活性; 在拉曼光谱中显示为强谱带的氰基在红外光谱中也没有活性; 在红外光谱中为弱吸收的苯环骨架振动, 在拉曼光谱中显示为强谱带, 这些都体现了红外光谱和拉曼光谱互补的优越性, 两种光谱相结合, 更有利于化合物结构的鉴定与检测。 然后通过实验方法测定了氯氰菊酯粉末的自然拉曼光谱, 将理论计算结果误差由频率校正因子0.973进行修正, 将实验结果与理论计算结果进行比较分析, 峰值频率波数相差大多在4~10 cm-1范围内, 理论与实验结果基本一致。 该研究为氯氰菊酯的振动光谱检测与结构鉴定提供了理论基础, 为其在农药检测领域的应用提供了理论参考。
As a broad-spectrum insecticide, Cypermethrin is widely used in various agricultural products, such as fruits, vegetables and poultry and so on. Because of its large dosage and slow degradation rate, drug residues in fruits, vegetables, livestock and other agricultural products are harmful to human health. In order to avoid human intake, it is very important to detect cypermethrin residues in agricultural products. Among the current detection methods, the vibration spectrum technology has the advantages of being non-destructive and fast. Therefore, this paper uses the density functional theory method combined with the vibration spectrum technology to provide a theoretical basis for the vibration spectrum detection and identification of Cypermethrin, and provide a reference for the application field of pesticide residue detection. The specific research contents and results are as follows: the first step is to construct the molecular space configuration of Cypermethrin by using Gaussian view software. Based on the DFT/B3LYP method of density functional theory, the structure is roughly optimized with a 3-21G basis set and then reoptimized with 6-311++G basis set based on coarse structure to obtain the stable configuration and frontier orbital distribution of the molecule. Then, based on the optimized structure, the theoretical infrared and Raman spectra of Cypermethrin were calculated. The theoretical results show that Cypermethrin has obvious infrared activity in the range of 3 300~3 000 and 1 700~500 cm-1. The former is mainly the vibration of functional groups, and the latter is the vibration of the fingerprint region. It can also be seen from the calculation results that the stretching vibration and scissor vibration of methylene hydrocarbon on cyclopropyl at 3 044 and 1 459 cm-1, the wagging vibration of methyne on cyclopropyl at 1 196 cm-1and the rocking vibration of hydrocarbon in benzene ring at 1 153 cm-1in Raman spectrum have no activity in the infrared spectrum. The cyano group without infrared activity shows a strong band in the Raman spectrum. The benzene ring skeleton vibration is weakly absorbed in the infrared spectrum but shows a strong band in the Raman spectrum. These reflect the complementary advantages of infrared spectroscopy and Raman spectroscopy. The combination of the two spectra is more conducive to the identification and detection of compound structure. In the second step, the natural Raman spectrum of Cypermethrin powder was measured by experimental method. The theoretical calculation error was corrected by the frequency correction factor of 0.973. The experimental results were compared with the theoretical calculation results. The difference in the peak frequency wavenumber was mostly in the range of 4~10 cm-1, and the theoretical data were consistent with the experimental results. This study provides a theoretical basis for the vibration spectrum detection and structure identification of Cypermethrin, and provides a theoretical reference for its application in pesticide detection.
氯氰菊酯(alpha-cypermethrin, CPM), 为白色或乳白色晶体或粉末。 熔点为78~81 ℃, 常温下几乎不溶于水, 易溶于醇类、 丙酮和芳烃等有机溶剂, 其化学名称为(1R, S)-顺, 反-2, 2-二甲基-3-(2, 2-二氯乙烯基)-环丙烷羧酸-(R, S)-α -氰基-3-苯氧基苄基酯, 化学式为C22H19Cl2NO3, 分子结构如图1[1, 2]。 氯氰菊酯又称灭百可、 兴棉宝等, 是一种含有氰基的Ⅱ 型拟除虫菊酯类仿生农药。 由于氯氰菊酯具有高效的广谱杀虫活性、 良好的稳定性及药效持久性等特点被广泛用于粮食、 果蔬等农产品的害虫防治[3]。 近年来研究发现氯氰菊酯对小鼠的生殖系统、 免疫系统、 肝脏等有危害, 对哺乳动物的中枢神经系统、 内分泌系统也存在显著危害, 并且有间接致癌作用[4, 5, 6, 7, 8, 9]。 由于氯氰菊酯具有用量大、 降解速度慢等特点, 其在使用过程中残留于农产品及环境中, 对人类健康造成不利影响。 对农产品及环境中的氯氰菊酯进行检测具有十分重要的意义。
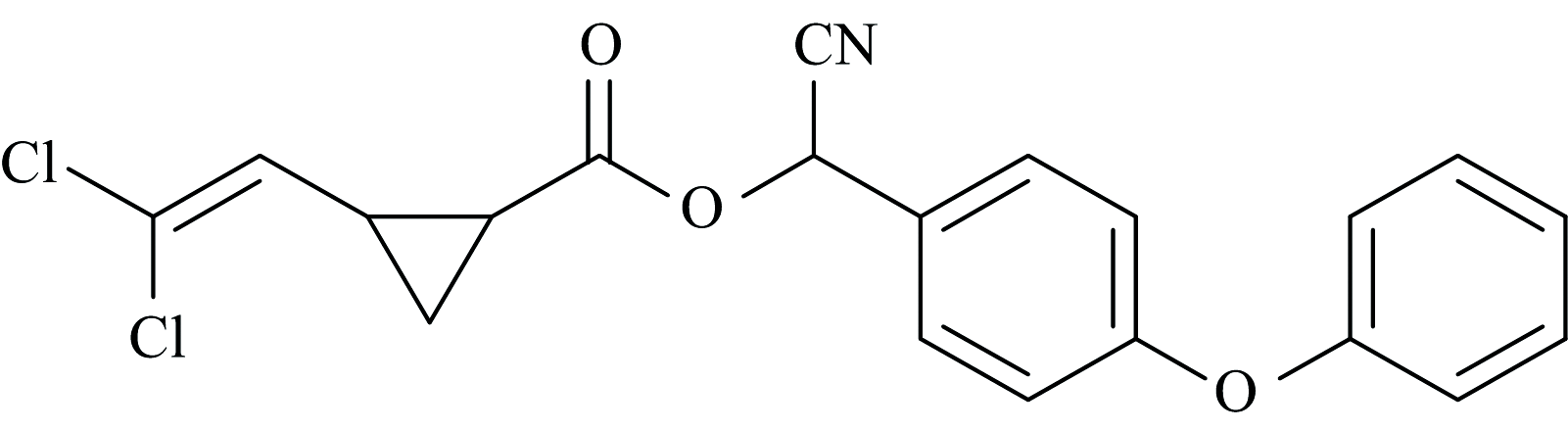 | 图1 氯氰菊酯分子平面结构Fig.1 Planar structure of cypermethrin |
红外光谱与拉曼光谱具有检出限度低、 无损快速, 并且光谱信息互补等优点, 在食品、 农产品及环境检测中得到广泛应用。 密度泛函理论(density functional theory, DFT)是一种研究多电子体系电子结构常用的量子力学方法, 能够直观地反映分子振动信息, 近年来被广泛用于研究反应机理、 振动光谱等[10]。 本工作采用密度泛函理论, 选择杂化密度泛函B3LYP方法, 对CPM化合物分子进行优化计算。 在优化结构的基础上, 对CPM进行了振动频率的计算, 并绘制了红外光谱和拉曼光谱图, 对两种谱图进行分析讨论, 同时对CPM固体粉末进行了实验拉曼光谱的测定, 将实验结果与理论计算结果进行比较分析, 为CPM的分析检测提供理论依据。
选用阿拉丁试剂官网的分析纯试剂氯氰菊酯, 按C22H19Cl2NO3计, 含量99%。 拉曼光谱测试采用DXR RamanMicroscope型拉曼光谱仪, 选择波长532 nm激光作为激发光源。
CPM的理论计算采用Gaussian09软件包, 分子构型由Gaussian view6.0构建。 由于CPM分子结构主要由C、 H、 O、 N等元素构成, 而基于密度泛函DFT方法的Beckes三参数混合模B3LYP泛函在轻元素构成的分子计算中被广泛应用, 并且计算得到的分子结构和光谱数据等与实验数据吻合较好, 比基于波函数的一些其他方法更为简单[10, 11, 12, 13]。
首先采用DFT/B3LYP方法, 在3-21G基组水平上, 对CPM分子进行几何结构粗优化, 在得到粗优化结构的基础上, 选择B3LYP/6-311++G基组进行再优化, 并基于优化后的结构计算分子的红外与拉曼光谱。 通常计算把所有振动都看作简谐振动而使计算频率较实验值偏大, 因此理论光谱的频率修正选择文献报道的0.973作为校正因子[14]。
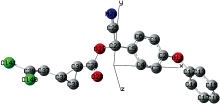
采用DFT/B3LYP方法, 在Gaussian09软件中优化后的CPM分子为三维非平面结构, 如图2所示。 CPM分子的结构主体为苯氧基苯, C23上连有一个乙腈基和羧基, C28上连接一个环丙基, C32上连接一个二氯乙烯基。 将CPM分子优化后的空间结构参数, 包括键长、 键角和二面角等列于表1。
 | 图2 B3LYP/6-311++G优化后的氯氰菊酯分子立体构型Fig.2 Optimized configuration of cypermethrin at B3LYP/6-311++G levels |
| 表1 优化后氯氰菊酯分子的键长、 键角和二面角数据 Table 1 Bond lengths and dihedral angles of cypermethrin obtained by configuration optimization |
表1的键角数据中, 环丙基上的三个角∠C30— C31— C32, ∠C31— C32— C30, ∠C32— C30— C31分别为61.534 95° , 59.589 19° 和58.875 86° , C1和C12所属苯环中的键角均在118° ~121° 之间, 说明CPM分子中的各碳原子和氧原子都是sp2杂化方式成键, 计算结果较理想。 从二面角数据∠C1— C2— O11— C12为167.523 64° , ∠C2— O11— C12— C13为-69.183 62° , ∠C3— C2— O11— C12为-14.197 95° , 可知CPM分子中苯氧基苯不是平面几何结构; 二面角∠C6— C5— C23— C25=-39.112 78° , ∠C4— C5— C23— C25=142.889 05° , 说明乙腈基与苯环不在同一平面。 表1中的键长数据显示CPM分子中的环丙基键长分别为: C30— C31为0.152 08 nm、 C30— C32为0.155 03 nm, 二者均比未取代的环丙烷键长0.150 11 nm长, C31— C32键长为0.150 96 nm, 则比未取代的环丙烷键长短, 说明环丙烷基团受取代基的影响产生了结构上的扭曲; C1和C12所属苯环中的碳碳键长较未取代苯环的键长也有所变化, 二者相比C1所在苯环的键长变化更显著, 说明取代基对苯环结构的影响较大。
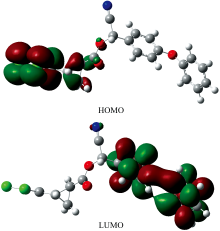
用DFT/B3LYP方法, 在6-311G++基组水平上优化得到的CPM分子的能量为-1 971.896 56 hartree, 即-53 655.305 37 eV, 优化构型能量较低。 偶极矩为6.165 73 D, 整个分子为极性分子。 计算得到的CPM分子的最高占有轨道(HOMO)和最低空轨道(LUMO)的电子云分布如图3所示。 HOMO能量为-8.131 71 eV, LUMO能量为-5.054 53 eV, 能隙为3.077 18 eV, 能隙较小, 离域π 电子容易被激发, 从HOMO和LUMO电子云分布图中也可看出。
 | 图3 氯氰菊酯分子的HOMO和LUMO图Fig.3 Highest occupied molecular orbital and lowest unoccupied molecular orbital of cypermethrin molecule |
2.3.1 CPM分子的红外光谱分析
红外光谱属于吸收光谱, 其特征性强, 提供的信息多, 从有机化合物的红外光谱可以得到丰富的结构信息, 因而在分析检测技术中广泛应用。 图4为采用DFT-B3LYP/6-311G++方法计算出的CPM分子的红外振动光谱。 与大多数的有机分子相同, CPM分子也具有不完全对称性, 在拉曼光谱和红外光谱中都有所体现, 通过Gaussian view 6.0观察CPM分子的理论红外光谱的振动形式, 对其振动模式归属进行指认。
 | 图4 氯氰菊酯分子的红外谱Fig.4 IR spectrum of cypermethrin |
首先, CPM的红外光谱中有2个极强吸收峰, 分别出现在1 130 cm-1为环丙基骨架振动引起, 1 218 cm-1为对位取代苯环的面内变形振动吸收峰。 其他特征吸收峰分析如下:
在859、 830和782 cm-1处的3个强吸收峰, 是芳环上H的面外变形振动, 指示了对位取代的C1苯环; 1 586~1 489 cm-1区域内有3个强吸收峰, 分别是两个苯环的骨架振动, 是由苯环伸缩引起的振动, 其中1 600 cm-1峰一分为二, 说明苯环与取代基产生了共轭; 3 200~3 000 cm-1区域的弱吸收峰, 为苯环上碳氢键的伸缩振动特征吸收带, $\nu=CH$出现在较高波数处, 说明苯环上的取代基起到了吸电子作用。
1 410 cm-1的强吸收峰表征环丙基中亚甲基的对称变形振动和两个次甲基的变形振动; 1 343 cm-1的强吸收峰, 是C30、 C32和C37所在的3个次甲基的面内变形振动; 997和798 cm-1处的2个强峰, 是环丙基的骨架振动吸收峰; 1 615 cm-1处的强吸收峰, 是羰基C38=O29的伸缩振动峰; 1 642 cm-1处的弱吸收峰, 是C37=O39双键的伸缩振动峰[15]。
2.3.2 CPM分子的拉曼光谱分析
拉曼光谱与红外光谱一样, 也是测定分子振动和转动的光谱, 但与红外光谱不同的是, 拉曼光谱属于散射光谱。 图5为采用DFT-B3LYP/6-311G++方法计算出的CPM分子的拉曼光谱。 通过Gaussian view 6.0观察CPM分子的理论拉曼光谱的振动形式, 对其振动模式归属进行指认。
 | 图5 氯氰菊酯分子的理论拉曼谱Fig.5 Theoretical Raman spectrum of cypermethrin |
与红外光谱相比, 拉曼光谱对结构的变化更敏锐, 一些在红外光谱中为弱吸收或强度变化的谱带, 在拉曼光谱中可能为强谱带, 更有利于对其进行检测[15, 16]。 例如苯环上碳氢键的伸缩振动特征吸收带在两种光谱中都位于3 300~3 000 cm-1区域, 显然由图4、 图5可见在拉曼光谱中呈强谱带, 在红外光谱中呈弱谱带; C37=O39双键的伸缩振动在红外光谱中为弱吸收峰, 而在拉曼光谱中则为强峰, 其红外吸收波数和拉曼位移均位于1 642 cm-1; C1=C2所在苯环的伸缩振动在红外光谱中为弱吸收峰, 而在拉曼光谱中则为强峰, 其红外吸收波数和拉曼位移均位于1 602 cm-1。 也有一些在红外光谱和拉曼光谱中强度相近的谱峰, 如1 615 cm-1处 C38=O29的伸缩振动峰, 在红外谱图和拉曼谱图中均有体现, 并且强度相近; 1 409 cm-1处的谱峰, 在红外谱图和拉曼谱图中的强度也相近。
有一些官能团的振动在红外光谱中能观察到, 但在拉曼光谱中观察不到; 而另一些官能团的振动, 在红外光谱中观察不到, 在拉曼光谱中则能观察到。 例如氰基的C25≡ N26伸缩振动在红外光谱(如图4)中观察不到, 但在拉曼光谱(如图5)中则为强峰, 位于2 232 cm-1; 另有以下振动模式只有拉曼活性: 3 044 cm-1的振动峰为环丙基上亚甲基碳氢的伸缩振动; 1 459 cm-1的振动峰为环丙基上亚甲基碳氢的剪式振动; 1 196 cm-1的振动峰为环丙基中次甲基的摇摆振动; 1 153 cm-1的振动峰为苯环中碳氢的面内摇摆振动; 999 cm-1的振动峰为C12所在苯环的骨架振动。 显然, 将红外和拉曼两种光谱结合即可获得关于氯氰菊酯分子结构的丰富而完整的信息。
将理论计算得到的拉曼光谱与实验测得的拉曼光谱进行对比, 如图5与图6所示。 由图6可见, 实验中CPM的拉曼光谱谱峰数量较理论结果要多, 并且谱峰强度明显, 说明实验结果较好。 比较理论计算结果和实验拉曼光谱的振动频率和光谱线型, 发现大部分谱峰的峰位是一致的。 如实验拉曼光谱中C38=O29的伸缩振动峰位于1 611 cm-1, 理论计算结果则位于1 615 cm-1; 实验结果中C1=C2所在苯环的伸缩振动峰位于1 597 cm-1, 理论结果位于1 602 cm-1; 环丙基上亚甲基碳氢的剪式振动位于实验拉曼光谱中1 465和1 413 cm-1, 而理论结果位于1 459和1 409 cm-1; 实验结果中C12所在苯环的骨架振动峰位于993 cm-1, 而理论结果位于999 cm-1。
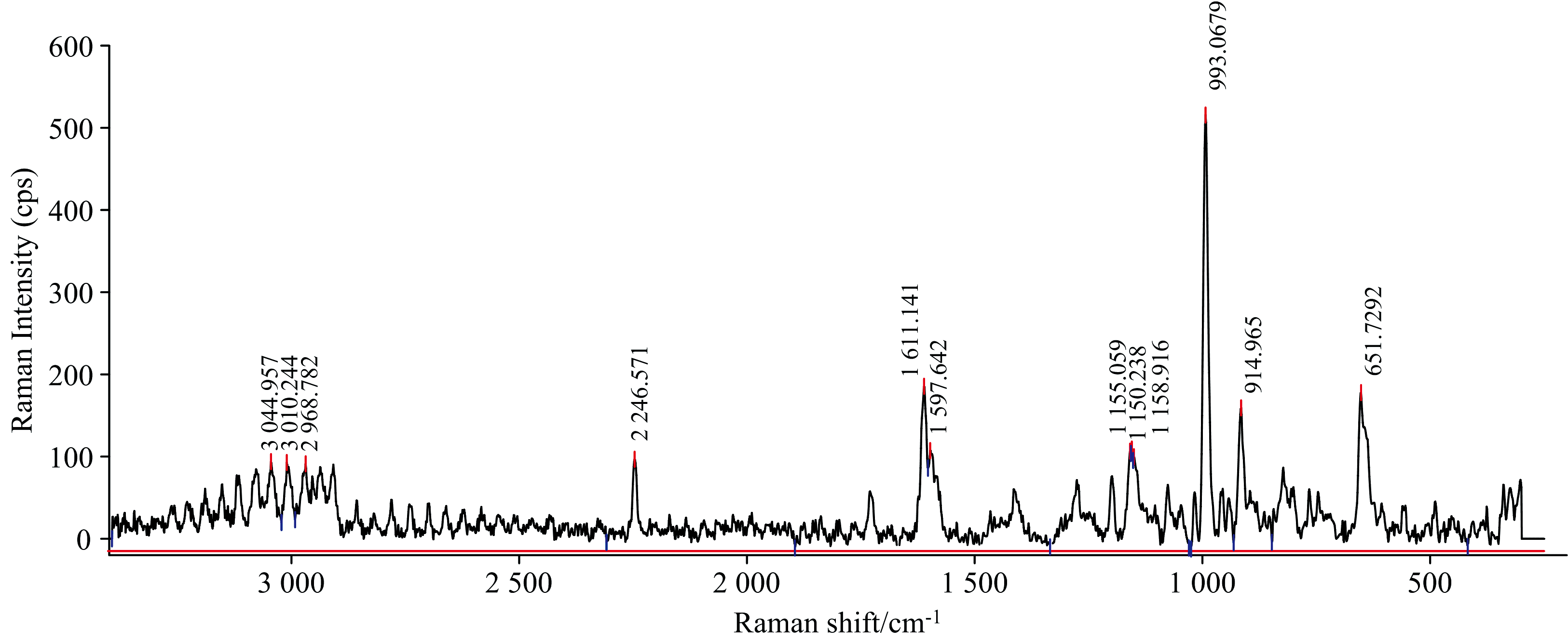 | 图6 氯氰菊酯分子的实验拉曼谱图Fig.6 Experimental Raman spectrum of cypermethrin |
实验和理论计算结果中的个别谱峰存在一定的差异, 这些差异主要体现在两个方面。 一方面是相应的峰位不一致, 另一方面是理论光谱中存在的个别谱峰在实验光谱中没有检测到。 这可能是由于实验设备产生随机误差, 同时理论计算模拟的是纯理论振动, 且DFT量子理论计算方法过多考虑了电子相关的影响, 而实验测定过程采用的是CPM的固体粉末, 存在分子间作用力的影响。
红外光谱和拉曼光谱是化合物鉴定和结构分析的两种重要手段。 红外光谱是由分子的不对称振动引起的振动-转动光谱, 属于分子吸收光谱, 而拉曼光谱是由具有对称分布的键的对称分布引起的, 属于散射光谱。 由于两种光谱有互补的特点, 二者配合使用可以得到化合物分子的完整振动信息, 对研究化合物结构十分有利。 本研究采用密度泛函DFT/B3LYP方法, 结合Gaussian09和Gaussian view6.0可视化软件, 首先对氯氰菊酯分子的结构进行了最低能量优化, 并计算得到了分子的前线轨道分布图, 讨论了分子最优的构型特点。 然后在优化构型的基础上, 选取6-311G++基组, 计算了氯氰菊酯分子的红外光谱和拉曼光谱, 确定了两种光谱各谱峰的振动模式归属, 并将两种光谱进行了对比分析。 同时对氯氰菊酯的分析纯样品进行了实验拉曼光谱的测定, 并将实验结果与理论计算结果进行了对比分析。 本研究结果可为除虫菊酯类农药的振动光谱检测和分子结构的鉴定提供光谱解析方面的理论依据, 为其在农药残留检测方面的应用提供理论参考。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|