



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TiO2-Ag纳米复合材料的制备及其光电催化性能研究
[陆燕华 , 徐敏敏, 姚建林
, 徐敏敏, 姚建林* ]
, 徐敏敏, 姚建林]
|
|


作者简介: 陆燕华, 1995年生, 苏州大学材料与化学化工学部硕士研究生 e-mail: 1043815274@stu.suda.edu.cn
载流子在等离激元金属纳米粒子上的快速复合, 导致传统的光电催化剂效率显著降低, 通过金属和半导体的复合可实现热电子和空穴的分离以提升光电催化效率。 采用Ag纳米粒子与半导体TiO2纳米粒子复合提高其光电催化活性, 并探索了催化活性提升的机理, 研究了TiO2-Ag纳米复合材料之间空间电荷区能带弯曲以及内置电场的作用, 为设计高性能SPR光电催化剂提供理论和实验依据。 以对氨基苯硫酚(PATP)及对硝基苯硫酚(PNTP)的光电催化偶联反应为探针, 研究了TiO2-Ag纳米复合材料的催化性能。 结果表明TiO2的引入提高了Ag的SPR催化活性, 其主要原因是TiO2的引入可提高TiO2-Ag间电子和空穴的分离效率。
The rapid recombination of carriers on plasmon metal nanoparticles significantly decreases the efficiency of traditional photocatalysts. The separation of hot electrons and holes can be achieved by recombining metal and semiconductors, improving the efficiency of photocatalysis. This paper combined Ag and TiO2 nanoparticles to improve the photocatalytic activity. The enhanced mechanism of the catalytic activity was explored. The effect of energy band bending in the space charge region between TiO2-Ag nanocomposites and the built-in electric field was studied, which provided a theoretical and experimental basis for designing high-performance SPR photocatalysts. Furthermore, the photocatalysis coupling reaction of PATP and PNTP was employed to study the catalytic performance of TiO2-Ag nanocomposites. The results reveal that the introduction of TiO2 improves the SPR catalytic activity of Ag. The main reason is that introducing TiO2 can improve the separation efficiency of electrons and holes between TiO2 and Ag.
近几十年来, 利用太阳的能量, 实现能源的可持续利用, 使得光催化成为清除污染的热门技术之一。 高于一定阈值的光照射到光催化剂上, 会产生电子和空穴对[1], 从而进一步催化降解污染物, 而该阈值则是半导体材料的本征特性。 已有报道的光催化剂中, TiO2由于合适的价带和导带位置可实现各种有机污染物的光催化降解[2], 因此成为研究最广泛的一类光催化剂。
有研究表明, 光催化技术可将多种有害污染物 (例如农药, 杀虫剂和芳香族有机物)降解并矿化成无害产品[3]。 该技术亦可用于还原重金属离子, 将有毒的重金属离子还原成不溶状态, 以便从工业废水中回收或去除[4]。 如Huang等发展了光催化技术, 使用TiO2从废弃物中将Ag+还原成Ag金属, 可同时实现水体污染处理和资源化利用[5], 利用TiO2增强荧光发光特性等, 有望进一步促进白光LED更加高效地应用。
然而, TiO2亦存在缺点, 如由于带隙较宽, 只能通过近紫外辐射激活光催化效应, 而近紫外辐射仅占自然光的4%~5%, 对太阳光的利用率较低; 光生电子-空穴对往往易于复合, 导致量子产率非常低[6]。 这些问题极大激发了科学家的研究热情。 已有报道通过将其吸收转移到可见光区域并防止电子和空穴的复合, 可增加TiO2光响应效率。 将离子掺杂到TiO2的晶格中实现可见光的响应[7], 由此缩小带隙改变TiO2的电子结构, 使掺杂的TiO2对可见光更加敏感, 或通过向催化剂中加载染料以及使用带隙较窄的半导体材料促使TiO2敏化, 使其可以被可见光激活[8]。 电子-空穴重新复合的问题, 已通过Pt, Ag或Au等金属对TiO2表面进行改性而得到改善[9]。 科学家们认为这些复合可使电子远离TiO2表面, 防止电子与空穴复合, 当金属功函数大于与其接触的半导体时, 形成肖特基势垒, 促进了电子从半导体到金属的转移。 提高催化效率的另一种途径是提高反应物在TiO2表面上的吸附。 以上两种途径可通过光催化剂表面沉积金属及修饰有机物同时实现[11]。
将Ag沉积于TiO2表面, 通过XPS技术研究TiO2-Ag之间的电荷转移, 以及电荷转移与肖特基势垒的关联。 利用空穴捕获剂, 研究内建电场与肖特基势垒对TiO2-Ag光催化效率的影响。
TiO2纳米粒子的合成过程与文献[11]相似, 将94.8 μ L正庚酸溶解在37.5 mL乙醇中, 将钛酸四丁酯(TBOT, 600 μ L)溶于3.6 mL乙醇中, 并迅速添加至剧烈搅拌的正庚酸乙醇溶液中。 在7.62 mL去离子水的作用下, 获得无定形TiO2胶体, 并在室温下剧烈搅拌2 h。 然后, 将胶体以6 000 r· min-1转速离心两次弃去上层清液, 用无水乙醇洗涤, 再转移至坩埚中60 ℃干燥。 将得到的粉末放入马弗炉中500 ℃退火2 h(升温速度为3 ℃· min-1)得到锐钛矿型TiO2 (100 nm)。
将上述制备所得0.1 g TiO2纳米粒子和1 g葡萄糖依次溶解, 配成40 mL水溶液[12]。 再将该混合液在60 ℃下搅拌10 min, 并在剧烈搅拌下迅速加入5 mL 0.4 mol· dm-3 AgNO3水溶液, 升温至90 ℃, 剧烈搅拌1 h, 确保Ag纳米粒子在TiO2表面的均匀吸附。 反应完成后, 将反应物自然冷却至室温, 以9 000 r· min-1的速度离心分离, 去除上层清液, 用超纯水和无水乙醇多次洗涤, 并在60 ℃下风干6 h, 所获得的灰色固体便是TiO2-Ag纳米复合材料。
GC电极在0.5 mol· dm-3 H2SO4溶液中, 控制电位为-1~1 V, 以0.1 V· s-1扫描速率进行40个循环清洗, 然后将5 μ L分散液(10 mg TiO2-Ag分散在5 mL水中)均匀分散在GC电极表面, 取出后用无水乙醇淋洗3次后晾干待用。
采用电化学粗糙的Ag电极作为工作电极进行对比研究。 Ag电极的粗糙步骤首先分别用金相砂纸对电极表面抛光以得到平整表面, 再分别用0.3和0.05 μ m的Al2O3粉末作为抛光粉, 将Ag电极表面多次长时间抛光, 在超纯水中超声清洗以去除电极表面残留的抛光粉。 将电极置于三电极电解池中, 选用0.1 mol· dm-3 KCl溶液作为电解液, 电位阶跃至+0.18 V并停留10 s进行氧化, 再以5 mV· s-1的扫速回至-0.25 V以还原电极表面。 得到表面呈淡黄色且具有SERS活性的粗糙Ag电极。
图1(a)是通过葡萄糖还原法制备的TiO2 (100 nm)-Ag纳米复合材料的透射电子显微镜(TEM)形貌图, 10~15 nm Ag纳米粒子均匀地分散在TiO2 (100 nm)的整个表面。
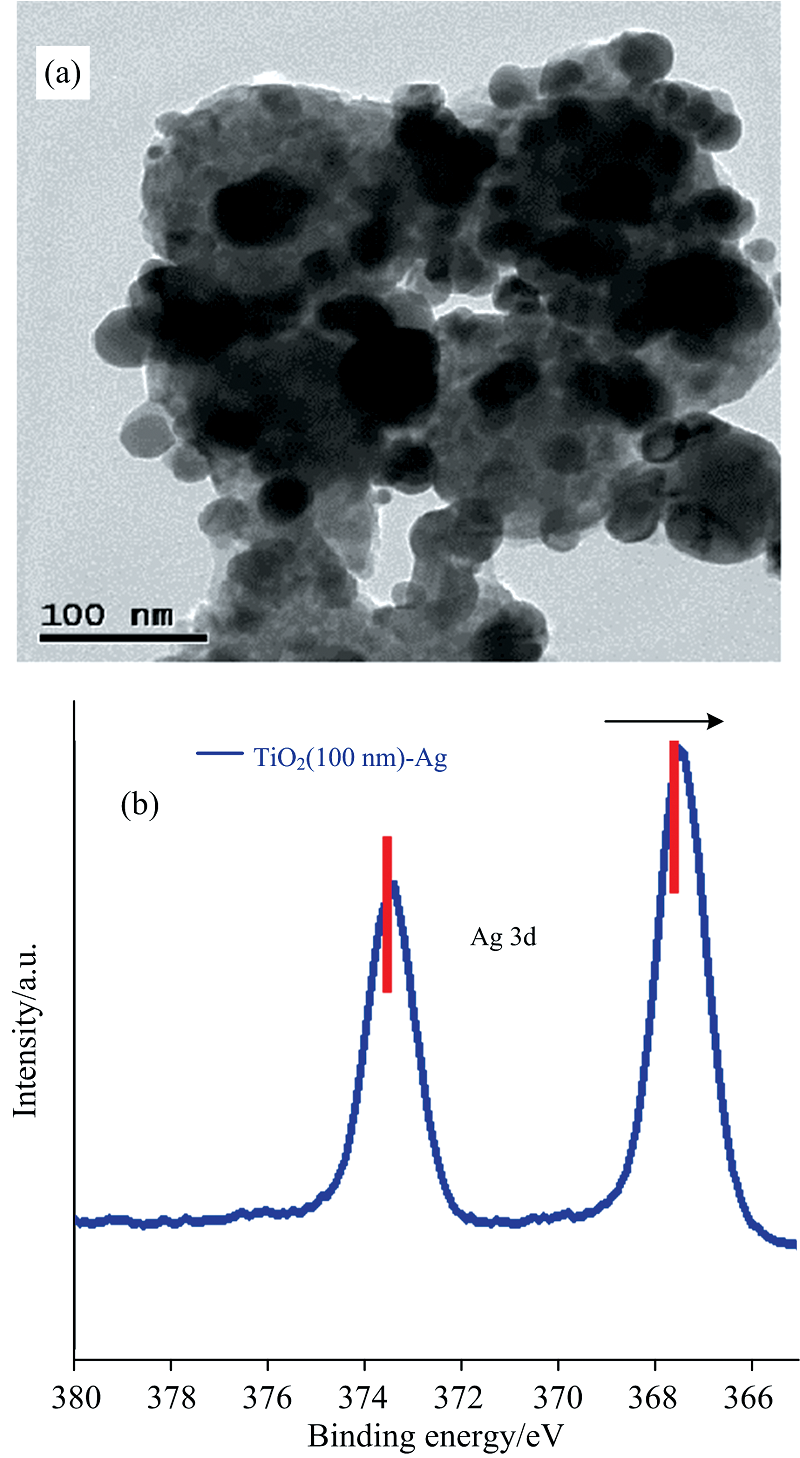 | 图1 TiO2 (100 nm)-Ag纳米复合材料的(a)TEM图和 (b)Ag(3d)的XPS区域谱光谱Fig.1 TEM image (a), and Ag(3d) XPS region spectrum (b) of TiO2 (100 nm)-Ag nanocomposites |
利用X射线光电子能谱(XPS)技术获得TiO2-Ag表面组成信息, 并依据284.5 eV的C(1s)标准结合能(BE)进行光电子峰校准。 图1(b)为TiO2表面Ag纳米粒子的XPS峰。 通过对比发现, TiO2 (100 nm)表面Ag纳米粒子的Ag 3d5/2和3d3/2峰发生了负移, 表明了TiO2-Ag纳米复合材料中Ag纳米粒子捕获电子的特征, 且TiO2 (100 nm)与Ag纳米粒子之间很容易发生电荷转移[13]。
以具有较好电荷转移效率的TiO2 (100 nm)-Ag作为催化剂, 选择对硝基苯胺(PATP)作为探针分子, 研究随着外加电位的变化, PATP向对二巯基偶氮苯(DMAB)转化的偶联过程。 根据文献报道在氧气环境中PATP偶联生成DMAB的吉布斯自由能为负值, 表明该反应是自发的放热反应[14]。 因此, 较正电位下立即出现的DMAB信号应该来自PATP在SPR催化下的自耦合过程。 为了获得对硝基苯硫酚(PATP)偶联效率的准确信息, 在测试开始时, 电位长时间保持在-0.9 V, 使得电极表面上所有激光照射下产生的DMAB都转化为PATP。 由于PATP的催化转化是可逆过程, 通过将电位正移至0 V, 获得随电位变化的偶联行为。 从图2(a)和(b)可见, 电位正移至-0.3 V时, 在1 148、 1 388和1 430 cm-1处出现了归属于DMAB的ag振动模式的谱峰[15]。 图3显示了在1 148和1 080 cm-1处两个峰的相对强度随电位的变化关系。 1 148 cm-1处的光谱峰属于DMAB的ag模式, 而1 078 cm-1是PATP和DMAB共有且具有相似散射截面的a1模式。 1 148与1 080 cm-1光谱峰的相对强度可以间接反映出PATP在电极表面的催化转化程度[16]。 即使在-0.9 V电位下, TiO2 (100 nm)-Ag电极表面仍有偶联发生。 而在均出现偶联的电位下, 如0 V时, TiO2 (100 nm)-Ag与粗糙Ag电极表面的偶联效率逐渐趋于一致。 这表明在极负电位下, TiO2的加入会促进Ag纳米粒子对PATP的催化偶联作用, 而在正电位下这种促进作用逐渐减弱, 在极端正电位下TiO2几乎不影响Ag表面的催化偶联性能。
 | 图2 在0.001 mol· dm-3 PATP和0.1 mol· dm-3 KCl的混合溶液中, PATP在(a)粗糙Ag电极和(b) TiO2 (100 nm)-Ag/GC电极表面随电位变化的SERS光谱Fig.2 Potential-dependent SERS spectra of PATP on roughened Ag electrode (a) and TiO2 (100 nm)-Ag/GC electrode (b)in the solution of 0.001 mol· dm-3 PATP and 0.1 mol· dm-3 KCl |
 | 图3 1 148和1 080 cm-1处SERS峰的相对强度随电位变化的分布图Fig.3 Potential-dependent Relative intensities profiles of the two peaks at 1 148 and 1 080 cm-1 |
为了研究TiO2-Ag的光催化增强机理, 选择乙醇作为空穴捕获剂[17], 以
 | 图4 (a)空穴捕获剂添加前后TiO2 (100 nm)-Ag/GC电极表面PATP在1 148和1 080 cm-1处SERS峰相对强度随电位变化的分布与(b)金属和n型半导体接触的能带Fig.4 (a) The potential-dependent relative intensities profiles of the two peaks at 1 148 and 1 080 cm-1 of PATP on TiO2 (100 nm)-Ag/GC electrode with or without hole capturing agent and (b) Energy band diagrams of metal and n-type semiconductor contacts |
当贵金属与n型半导体TiO2表面接触时, 如前者功函小于后者, 金属表面的电子会自发的向半导体表面流动且发生聚集产生电子积累层。 电子的这种自发流向, 势必引起TiO2费米能级的下降, 随即导致界面带的向下弯曲, 由此在界面形成欧姆接触[18]。 而由于实际情况下Ag (-4.7~-4.6 eV)与TiO2 (-5.0~-4.5 eV)的费米能级易受界面处氧气和水吸附等因素的影响, 导致界面处欧姆接触与肖特基势垒之间发生转化, 给催化反应机理研究增添了困难。 本实验相应的XPS研究中可知, TiO2 (100 nm)-Ag复合材料中电荷转移是自发的由半导体向Ag纳米粒子进行的, 因此该复合材料之间产生的势垒实际上仍为肖特基势垒[18]。 如图4(b)所示, 该肖特基势垒可以有效地作为电子陷阱, 以防止光生电子和空穴重新结合, 并延长光生电子和空穴对的寿命。 在空间区域中能带的弯曲和内置电场的存在促进了电子和空穴的分离, 并促进了诸如离子、 电子和空穴等载流子的扩散。 自由载流子密度和局部电导率的这种变化将不可避免地导致界面活性的增加。
以粗糙Ag电极作为工作电极, 0.001 mol· dm-3 PNTP和0.1 mol· dm-3 KCl作为反应液进行SERS测试, 在粗糙Ag电极表面, PNTP受SPR催化生成DMAB, 随后在-0.6 V的还原电位下向PATP发生转化[19, 20, 21, 22]。 1 574 cm-1附近归属于苯环的v(C=C)特征峰向1 597 cm-1发生蓝移, 谱图变化一直持续至电位负移到-0.9 V, 此时苯环上v(C=C)完全蓝移至1 597 cm-1, 谱图与PATP的SERS谱图相吻合, 说明随着电位的负移, 电极表面分子会完全还原为PATP。
还原电位下DMAB向PATP的转化会干扰对偶联反应效率的判断。 因此, 为了更好地探究TiO2 (100 nm)-Ag对PNTP偶联反应的影响, 首先将电位负移至-0.9 V, 使得电极表面分子完全还原至PATP, 再逐渐将电位从-0.9 V向0 V正移。 如图5(a)所示, 随着电位的正移, 归属于ν (NO2)的特征峰出现, 因为测试环境是PNTP的KCl溶液, 表明在正电位下PNTP再一次吸附于电极表面。 因此, 图5(a)谱图中出现的1574 cm-1处归属于苯环的v(C=C)特征峰为PATP偶联与电解液中的PNTP偶联共同贡献。 如图5(b)所示, 由1 142 cm-1与1 080 cm-1处光谱峰的相对强度比较可知[16], 在利于偶联反应进行的0 V电位下, 粗糙Ag电极表面的相对偶联效率达到了2.0, 同等条件下TiO2-Ag电极表面的相对效率为2.1, 仅略有提高。 而在不利于偶联反应的-0.9 V电位下, 粗糙Ag电极表面几乎不发生偶联, 而TiO2-Ag电极表面依旧能顺利进行偶联反应, 且相对偶联效率为0.35。 结果与图3的结论相一致, 在较负的还原电位下, TiO2的加入会促进Ag纳米粒子对偶联反应的催化作用, 而在相对较正的电位下这种促进作用逐渐减弱。
 | 图5 (a)在0.001 mol· dm-3 PNTP和0.1 mol· dm-3 KCl的混合溶液中, PNTP在TiO2 (100 nm)-Ag/GC电极表面随电位变化的SERS光谱; (b) 1 142和1 080 cm-1处SERS峰的相对强度随电位变化的分布Fig.5 Potential-dependent SERS spectra of PNTP on TiO2 (100 nm)-Ag/GC electrode in the solution of 0.001 mol· dm-3 PNTP and 0.1 mol· dm-3 KCl (a); potential-dependent relative intensities profiles of the two peaks at 1 142 and 1 080 cm-1 (b) |
进行了TiO2-Ag纳米复合材料促进光催化反应的机理研究。 通过葡萄糖还原法制备了TiO2 (100 nm)-Ag复合纳米结构, 研究了可见光照射下SPR催化性能。 采用XPS技术研究TiO2-Ag复合材料之间的电荷转移。 采用探针分子策略和SERS技术研究金属-半导体纳米复合材料中的界面电荷转移方向, 发现电子可自发从TiO2转移到Ag纳米粒子表面, 表明该还原法制备的TiO2-Ag纳米复合材料之间仍是肖特基势垒, 与制备的环境以及TiO2-Ag所受的外界干扰有关。 将优化的TiO2-Ag应用于经典SPR催化反应中, 发现能在负电位下显著提高偶联反应效率, 表明TiO2的加入能促进Ag表面SPR催化反应。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|