

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于外电场作用对二溴苯的光谱特性和解离特性
[安桓1  , 闫好奎
, 闫好奎2 , 向梅1, * , 布玛丽亚·阿布力米提1, * , 郑敬严1 ]
, 闫好奎, 布玛丽亚·阿布力米提, 郑敬严|
|


作者简介: 安 桓, 1994年生,新疆师范大学物理与电子工程学院硕士研究生 e-mail: anhuan7643@163.com
对二溴苯(C6H4Br2)在化学工业领域有着广泛的应用, 但也是威胁臭氧层的有机污染物之一, 研究外电场作用下该分子的解离特性对臭氧层的保护具有重要参考价值。 在不同外电场(-0.025~0.025 a.u.)作用下, 采用密度泛函理论(density functional theory, DFT), B3LYP/6-311+G(d, p)基组水平上优化了对二溴苯分子的基态几何结构; 采用含时密度泛函理论(time-dependent density functional method, TD-DFT)和B3LYP/6-311+G(d, p)基组计算了分子的紫外吸收光谱, 推测分子的解离特性; 对分子两个C—Br键的势能进行扫描, 给出了对二溴苯分子解离特性的直接证据。 研究表明, 在外电场作用下, 对二溴苯分子的基态几何结构、 光谱特性、 势能曲线及势能面均发生较大改变。 随着外电场的增加, 对二溴苯分子的3C—12Br键长、 分子体系总能量均逐渐降低, 6C—11Br键长、 偶极矩逐渐增大; 6C—11Br键长的增大, 说明对二溴苯分子的6C—11Br键能减小, 6C—11Br键更容易断裂。 能隙随外电场增强先增大后减小, 能隙的减小, 说明分子更容易发生化学反应。 C—Br键伸缩振动峰强度逐渐减小, 紫外吸收光谱吸收峰强度先略微增大后猛然降低, 红外光谱的振动频率和紫外吸收光谱的最强峰均发生了红移, 表现出分子能量增强的特性, 表明振动加强, 化学键变得更活跃。 在外电场作用下扫描了对二溴苯分子的3C—12Br键, 得到了分子3C—12Br键的势能曲线, 当外电场强度为-0.02 a.u.时, 分子右侧势垒的最高能量与最低能量基本相平, 分子3C—12Br键断裂; 在此电场强度下继续扫描6C—11Br的势能发现, 分子6C—11Br键也会断裂, 因此对二溴苯分子可以发生逐步解离。 在外电场作用下同时扫描对二溴苯分子的两个C—Br键, 得到分子的势能面, 当外电场强度为0.02 a.u.时, 势能面的对角线势能降低, 出现另一个解离通道, 因此对二溴苯分子可能发生协同解离。 上述结果为实验研究对二溴苯分子的外电场降解机理提供数据保障, 也对该分子体系的解离特性研究有重要的参考意义。
P-dibromobenzene (C6H4Br2) has a wide range of applications in the chemical industry, but it is also one of the organic pollutants that threaten the ozone layer. The study of the dissociation characteristic of the molecule under an external electric field has important reference value for protecting the ozone layer. Under the action of different external electric fields (-0.025~0.025 a. u.), the p-dibromobenzene molecule is optimized based on B3LYP/6-311+G(d,p) based on density functional theory (DFT), the ground state geometric structure is computed. They are then using the Time-dependent density functional theory (TD-DFT) and B3LYP/6-311+G(d,p) basis set to calculate the ultraviolet absorption spectrum of the p-dibromobenzene molecule to guess the dissociation property of the molecule. Finally, scanning the potential energy of the two C—Br bonds of the molecule gave direct evidence of the dissociation property of the p-dibromobenzene molecule. Studies have shown that under the action of an external electric field, the ground state geometric structure, spectral characteristics, potential energy curve and potential energy surface of p-dibromobenzene molecules have undergone major changes. With the increase of the external electric field, the 3C—12Br bond length and total energy of the molecular system of p-dibromobenzene gradually decrease, and the 6C—11Br bond length and dipole moment gradually increase; the increase of the 6C—11Br bond length indicates that the 6C—11Br bond energy of p-dibromobenzene molecule is reduced, and the 6C—11Br bond is the easiest to break. The energy gap first increases and then decreases with the increase of the external electric field. The decrease in the energy gap indicates that the molecule is more prone to chemical reactions. The peak intensity at the stretching vibration of the C—Br bond gradually decreases. The peak intensity at the stretching vibration of the C—Br bond gradually decreased, and the absorption peak of the ultraviolet absorption spectrum increased slightly at first and then decreased abruptly. The energy-enhancing properties indicate that the vibrations intensify, and the chemical bonds become active. The 3C—12Br bond of the p-dibromobenzene molecule was scanned under an external electric field, and the potential energy curve of the 3C—12Br bond of the molecule was obtained. When the intensity of the external electric field is -0.02 a.u., the highest energy of the right barrier of the molecule is the same as the lowest energy. Flat, the molecular 3C—12Br bond is broken; continue to scan the potential energy of 6C—11Br under this electric field strength, the molecular 6C—11Br bond is broken, so the p-dibromobenzene molecule will gradually decompose the separation phenomenon. Under the action of an external electric field, scan the two C-Br bonds of p-dibromobenzene simultaneously to obtain the molecule's potential energy surface. When the external electric field intensity is 0.02 a.u., the diagonal potential energy of the potential energy surface decreases, and another dissociation channel appears. Therefore, synergistic dissociation of p-dibromobenzene molecules may occur. These results provide data guarantee for the experimental study of the degradation mechanism of p-dibromobenzene molecules in an external electric field and have important reference significance for the study of the dissociation characteristics of the molecular system.
臭氧层作为地球的“宇航服”, 吸收短波紫外线和部分中波紫外线, 保护地球上的生物得以生存和繁殖。 1974年, 美国科学家Molina等阐述了关于氟利昂对臭氧层具有很大的破坏性[1]; 1985年, Farman等通过在南极站观测进一步证实此结论[2], 但对于臭氧层的破坏方式依旧存在争议。 2009年, 中国台湾科学家林志民团队研究ClOOCl在激光作用下的解离速率, 才以明确的证据平息[3]。 经科学研究发现, 一般在大气下层发生二体解离过程, 而在平流层等大气较高层, 经常会发生多体解离现象。 为了理清溴自由基的产生机理, 了解溴代化合物对大气环境的影响, 研究溴代化合物的解离动力学很有必要。
三体解离动力学因可提供解离过程的动态演化信息而受到研究者持续广泛的关注[4]。 根据时间差(τ)和待解离中间体碎片转动周期(Tr)的相对关系, 可将三体解离区分为顺序解离和协同解离; 根据τ/Tr和1的大小关系又可以将解离细分为顺序解离(τ/Tr>1)、 同步解离(τ/Tr=0)和非同步解离(0< τ/Tr< 1)。 目前主要通过实验方法来研究分子的协同解离现象, Pragya团队采用离子分子碰撞装置研究了OCS3+的三体解离, 结果显示能量为200 kV·q-1的Xe9+与OCS分子多次碰撞使分子发生协同解离和逐步解离[5]; 有报道通过飞行时间质谱技术研究了氟利昂1110在800 nm飞秒脉冲光作用下的多光子解离, 采用离子速度成像技术印证了C2Cl3+与C2Cl2+分别对应逐步解离和协同解离通道[6]; Lee等观察到丙烯醛在193 nm的同步辐射光源激发下, 探测到协同解离碎片C2H2+CO+H2, 并利用量子化学计算确定了基态势能面上的相应跃迁结构, 该研究为同步协同三体解离提供了新的机制, 并将光化学研究扩展到通过打破非等价化学键的同步协同多重解离[7]。 在理论方面也可以研究分子协同解离现象, 如Mackey等在密度泛函理论B3LYP/6-31G(d)基组水平上采用Cope重排研究了1, 5-己二烯的解离现象, 发现了95%的协调解离和5%逐步解离轨迹[8]; Duan等借助Gaussian计算软件通过密度泛函理论B3PW91/6-311+G(2df)基组计算了二氟二溴甲烷的势能面, 研究发现外电场强度为0.055 a.u.时两个C—Br键依次解离, 外电场强度为0.065 a.u.时协同解离现象发生[9]。
对二溴苯作为生产聚氨酯的中间体和合成药物的试剂被广泛应用于化学工业领域。 由于对二溴苯分子在紫外辐射下会生成溴自由基, 与大气中的臭氧层发生连锁反应(Br+O3→ BrO+O, BrO+O3→ Br+2O2), 造成对臭氧层的破坏, 因此对二溴苯的降解问题引起了学者的关注。 对二溴苯分子常用的降解方法有光催化降解[10]、 电化学降解[11]等。 在外电场作用下, 分子会发生一系列物理和化学变化, 例如分子化学键断裂, 分子轨道、 晶体感应、 激发光谱等发生变化[12, 13]。 采用外电场降解此类污染物是一种较实用的新思路, 已应用于哈龙分子和甲烷衍生物等分子的降解机理的研究[14], 相较其他方法, 外电场降解所需的应用条件较低, 便于工业化应用。 基于外电场作用理论研究对二溴苯分子, 不仅可以得到分子的基态几何性质、 光谱特性及势能面等信息, 还由于对二溴苯分子具有高度对称性, 有两个等价的C—Br键, 可以尝试观察其协同解离现象。 因此研究外电场作用下对二溴苯分子的光谱特性和解离特性很有意义。 目前采用外加电场理论研究对二溴苯分子的逐步、 协同解离还鲜有报道。
本工作采用密度泛函理论B3LYP/6-311+G(d, p)基组研究了对二溴苯分子的基态几何构型、 光谱特性和解离特性, 为对二溴苯外电场降解提供数据保障。 首先优化了对二溴苯的基态几何构型, 包括键长、 能量、 偶极矩、 能级分布等物理参数。 采用含时密度泛函理论在B3LYP/6-311+G(d, p)基组水平上研究了对二溴苯分子的紫外吸收光谱。 采用相同方法和基组计算了分子C—Br键的单点扫描能量, 得到了单键的势能曲线和双键的势能曲面。 研究对二溴苯分子在外电场作用下的解离特性, 可帮助了解分子在解离过程中的迁移规律, 为提高对二溴苯的降解效率提供了重要参数。
在外电场作用下对二溴苯分子体系哈密顿量H为
式(1)中, H0为无外场时的哈密顿量, Hint为外电场与分子体系的相互作用的哈密顿量。 在偶极近似下, 分子体系与外电场F的相互作用能为
式(2)中, μ为分子的电偶极矩, F为点电荷模型或者有限场模型下的电场强度, 其中1 a.u.=5.142 25×1011 V·m-1(a.u.为原子单位)。
根据Grozema等提出的模型[15], 在外电场作用下的激发能Eexc与电场强度F, 电偶极矩和极化率的变化量Δu和Δα满足如式(3)关系
式(3)中, Eexc(0)为零电场时的激发能。 吸收振子强度flu为
式(4)中, 线强度S为原子单位(e2
通过不同的方法和基组模拟多种情形, 对对二溴苯分子进行结构优化计算, 将计算得到的键长、 键角与实验值作比较, 选取出最合适的密度泛函理论B3LYP/6-311+G(d, p)基组; 通过加不同电场, 计算并分析了对二溴苯分子的几何构型、 偶极矩、 电荷分布、 振动频率、 轨道能级分布、 红外光谱、 解离势能面以及分子紫外吸收光谱的变化规律。 理论计算均采用Gaussian 09[16]量子化学计算软件进行。
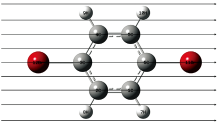
在无外电场作用下, 对对二溴苯分子不同方法和基组进行优化计算, 将优化出的平衡核间距R、 键角A与实验值做比较, 结果如表1所示。 密度泛函理论B3LYP/6-311+G(d, p)基组得到的平衡核间距、 键角与实验数据基本相符, 因此, 沿着箭头所示方向加外电场时均选用DFT/B3LYP方法6-311+G(d, p)基组进行计算。 分子基态几何结构与外加电场方向如图1所示。
| 表1 不同方法优化对二溴苯分子基态结构参数 Table 1 Different methods to optimize the ground state structure of p-dibromobenzene molecule |
 | 图1 无外电场下的对二溴苯分子稳定基态构型Fig.1 The stable ground state configuration of p-dibromobenzene molecule without an external electric field |
采用密度泛函理论在B3LYP/6-311+G(d, p)基组水平上沿箭头方向(X轴)加不同外电场(-0.025~0.025 a.u.)对分子进行结构优化, 得到分子稳定构型, 对二溴苯分子的键长、 电荷布局数如表2所示。 数据表明, 外电场作用下分子的结构发生了明显的改变。
| 表2 不同外电场作用下对二溴苯分子的几何结构参数 Table 2 The geometric structure of p-dibromobenzene molecules under different external electric fields |
键长的变化如图2所示, 分子3C—12Br键长随外电场增强而减小, 6C—11Br键长随外电场增强而增大。 分子键长随外电场的变化规律可以用分子内部电场的变化来解释: 当有外电场存在时, 分子内的电子将向与外电场相反的方向转移, 平衡结构由分子内应力和外电场应力的组合决定。 研究结果表明, C—Br键能量最高, 且外加电场越大, 6C—11Br键能量越低, 即6C和11Br核间距增大, 6C—11Br键越容易断裂。 6C和11Br原子间净电荷量如表2所示, 随外电场强度净电荷量均逐渐增强, 说明6C和11Br原子间库仑引力增大, 6C—11Br键长增大; 而3C和12Br原子间作用力反之。
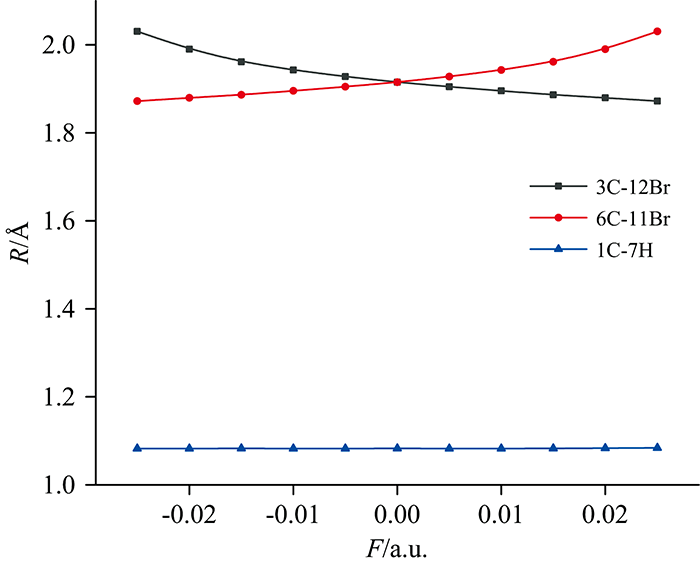 | 图2 不同外电场作用下对二溴苯分子键长的变化Fig.2 The change of p-dibromobenzene molecular bond length under different external electric fields |
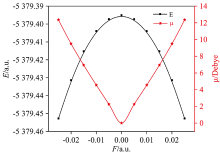
不同外电场下分子总能量和电偶极矩的变化如图3所示, 总能量随外电场增强先增大后减小, 而电偶极矩随外电场增强先减小后增大, 表现出很好的对称性。 分子体系和外电场的相互作用能为Hint=-μF, 分子系统能量H的哈密顿势能先减小后增加, 使分子体系与外电场的相互作用能减小, 也可以解释分子体系的总能量随外电场的增强而减小的现象。
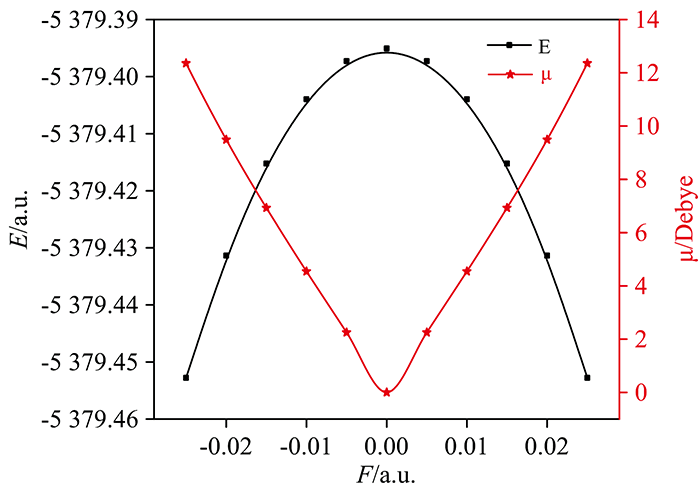 | 图3 不同外电场作用下对二溴苯分子 总能量、 偶极矩的变化Fig.3 The change of p-dibromobenzene molecular total energy and dipole moment under different external electric fields |
不同外电场(-0.025~0.025 a.u.)作用下, 分子的最低空轨道(LUMO)能量EL和最高占据轨道(HOMO)能量EH以及对能隙EG的影响如表2所示, 其中能隙EG计算公式为式(5)
分子前沿轨道能决定许多分子性质, 在物理和化学中具有重要意义。 EL表示分子得到电子能力的强弱, EL越小越容易得到电子; EH表示分子失去电子能力的强弱, EH越大越容易失去电子; EG表示分子参加化学反应的能力, EG越小分子越容易被激发到激发态而发生化学反应。 从图4可以看出, 随着外电场的增强EL先增大后减小, EH先减小后增大; 能隙随着外电场增强先增大后减小, 说明电子越容易被激发到空轨道而形成空穴子, 从而发生化学反应, 与2.2节6C—11Br键在外场作用下发生断裂的结论一致。
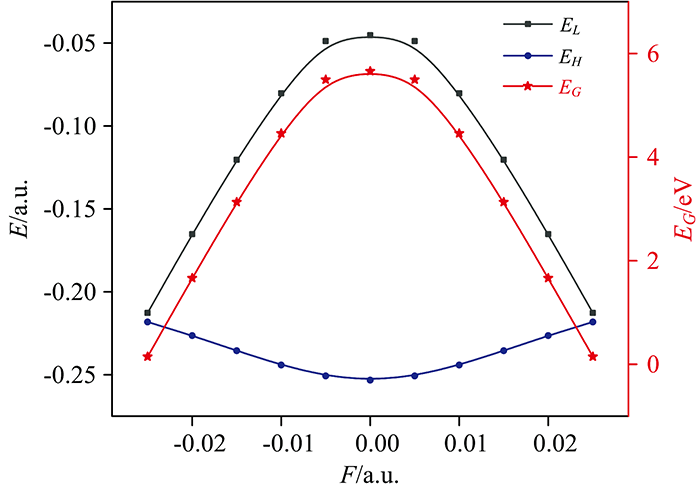 | 图4 不同外电场作用下对二溴苯分子最低空轨道能、 最高占据轨道能和能隙的变化Fig.4 The change of p-dibromobenzene molecular lowest empty orbital energy, the highest occupied orbital energy and energy gap under different external |
研究分子的光谱特性, 对了解分子的性质以及结构具有重要意义。 沿着箭头方向(X轴)加不同外电场(-0.025~0.025 a.u.), 采用合适的方法在B3LYP/6-311+G(d, p)基组水平上对分子进行结构优化, 得到对二溴苯分子稳定构型。 由红外和紫外吸收光谱可以看出, 外电场作用下分子的光谱特性发生了明显的变化。
2.4.1 外电场作用下对二溴苯的红外光谱
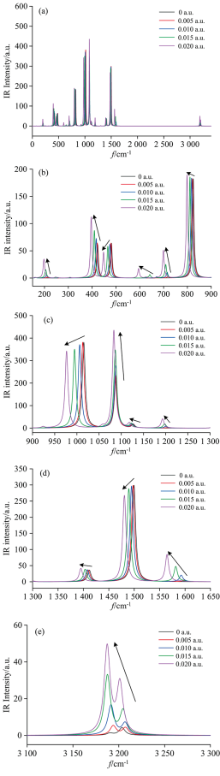
C—Br键拉伸振动的变化对于研究外加电场作用下对二溴苯的解离机理具有重要意义。 研究表明, 从红外光谱以及振动频率可以看出, C—Br键的伸缩振动主要位于423.15, 719.74, 1 015.52, 1 074.76, 1 087.67, 1 499.53以及1 602.32 cm-1处, 但是423.15, 719.74, 1 015.52和1 074.76 cm-1四处C—Br键的伸缩振动幅度较大, 集中了主要能量; 1 087.67, 1 499.53及1 602.32 cm-1处C—Br键的伸缩振动幅度较小, 能量主要集中在苯环的振动。
外电场的作用下, 对二溴苯分子的红外光谱如图5(a)所示。 为更清楚观察其规律, 将红外光谱分块放大。 如图5(b—e)所示, 随外电场的增强, 分子所有振动频率f均减小, 由公式λ=u/f可知(其中u为波速, f为频率, λ为波长), 分子的波长λ增大, 该分子所有振动频率均发生了红移, 同时C—Br键伸缩振动峰强度逐渐减小。
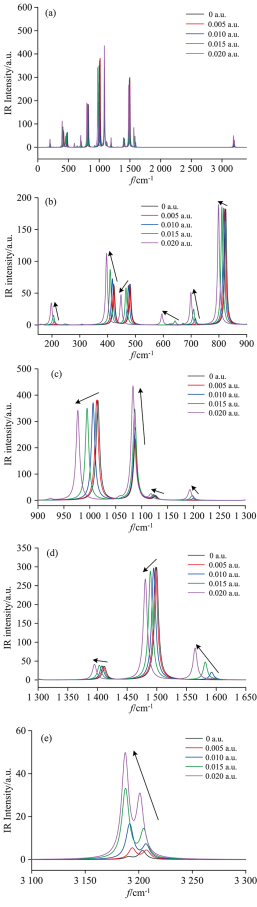 | 图5 不同外电场作用下对二溴苯分子红外光谱的变化 (a): 全谱图; (b): 150~900 cm-1; (c): 900~1 300 cm-1; (d): 1 300~1 650 cm-1; (e): 3 100~3 300 cm-1Fig.5 The change of p-dibromobenzene molecular the IR spectrum under different external electric fields (a): Infrared spectra of all frequncies; (b): 150~900 cm-1; (c): 900~1 300 cm-1; (d): 1 300~1 650 cm-1; (e): 3 100~3 300 cm-1 |
2.4.2 外电场作用下对二溴苯的紫外吸收光谱
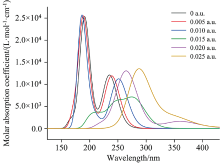
沿着箭头方向(X轴)加不同外电场(-0.025~0.025 a.u.), 采用含时密度泛函理论在B3LYP/6-311+G(d, p)基组水平上对分子进行结构优化, 得到对二溴苯分子稳定构型。 对二溴苯分子的紫外吸收光谱如图6所示, 随着外电场的增强, 对二溴苯分子紫外吸收光谱的吸收峰波长发生了红移, 峰值先略微增大, 后猛然降低, 峰值约为前者的一半; 峰形变宽, 但是所有变化均在紫外波段。 实验表明, 正向外电场的增强, 使得电子态的跃迁能降低。
 | 图6 不同外电场作用下对二溴苯分子紫外吸收光谱的变化Fig.6 The change of p-dibromobenzene molecular the UV-Vis under different external electric fields |
研究外电场作用下对二溴苯分子的解离特性是工作的关键。 沿着图1所示箭头方向(X轴)加不同外电场(-0.025~0.025 a.u.), 采用密度泛函理论在B3LYP/6-311+G(d, p)基组水平上对分子进行结构优化, 得到对二溴苯分子稳定构型。 数据表明, 外电场作用下分子的势能曲线、 势能面发生了明显的变化。
2.5.1 外电场作用下对二溴苯的逐步解离
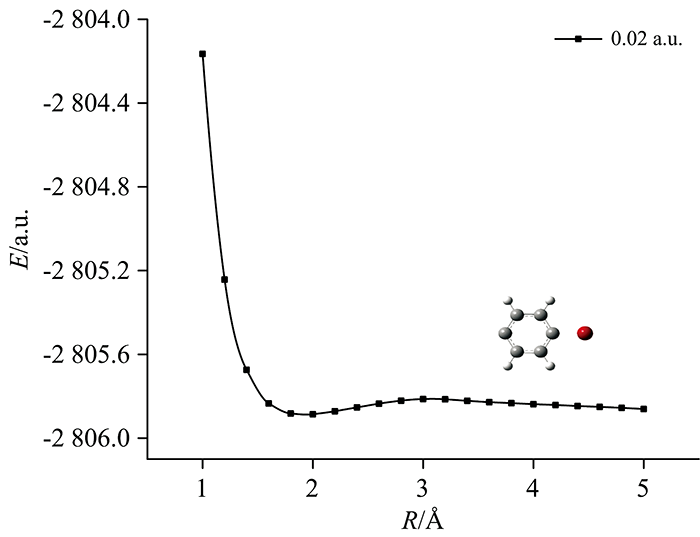
加外电场对分子两个C—Br键的最低能量进行扫描, 其中C和Br原子的核间距为1~5 Å , 每步为0.2 Å , 得到外电场作用下对二溴苯分子的势能曲线如图7和图8所示。
 | 图7 不同外电场作用下对二溴苯分子3C—12Br的势能曲线Fig.7 The potential energy curve of p-dibromobenzene molecular 3C—12Br under different external electric fields |
 | 图8 不同外电场作用下对二溴苯分子6C—11Br的势能曲线Fig.8 The potential energy curve of p-dibromobenzene molecular 6C—11Br under different external electric fields |
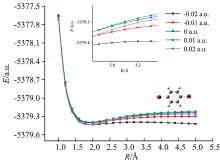
外电场作用下对二溴苯分子3C—12Br的势能曲线如图7所示, 无外电场时, 对二溴苯分子能量先急剧下降, 达到稳定, 后随着3C与12Br原子的核间距增大而适度升高, 使得分子处于两个较大势垒的稳定点。 增加外电场时, 分子随外电场的增强, 右侧势垒逐渐下降, 说明3C—12Br键可能会断裂。 当外电场强度为-0.02 a.u.时, 分子右侧势垒的最高能量与最低能量基本相平, 说明3C—12Br键断裂, 分子发生解离。
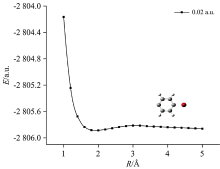
保持外电场强度为0.02 a.u., 继续扫描6C和11Br原子间势能, 得到外电场作用下C6H4Br分子6C—11Br的势能曲线如图8所示。 从图可以看出, 右侧势垒很低, 说明在该外电场作用下分子的6C—11Br键不稳定, 会发生断裂。 综上所述, 在外电场作用下对二溴苯分子可以发生逐步解离。
2.5.2 外电场作用下对二溴苯的协同解离
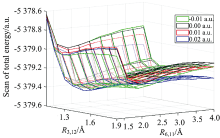
为了解对二溴苯分子在外电场作用下的协同解离现象, 采用密度泛函理论在B3LYP/6-311+G(d, p)基组水平上对分子的两个C—Br键同时进行扫描。 首先固定分子一个C—Br键为1.3 Å , 扫描另一个C—Br键从1.3 Å 至4 Å , 步长设置为0.3 Å ; 接着固定分子一个C—Br键为1.3 Å , 扫描另一个C—Br键从1.3 Å 至4 Å , 步长设置为0.3 Å 。 重复这个过程并收集数据, 得到外电场作用下对二溴苯分子两个C—Br键的势能面如图9所示, 对二溴苯分子在零点场下处于束缚态。 随着外电场增强, 势能面逐渐降低; 6C和11Br的核间距减小, 出现一个解离通道。 当外电场强度为0.02 a.u.时, 对角线方向势能降到最低, 说明两个键可能同时断裂, 对二溴苯分子可能出现解离通道现象。
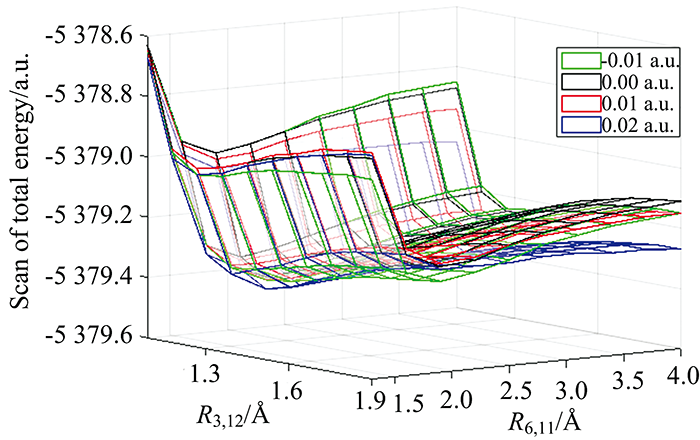 | 图9 不同外电场作用下对二溴苯分子的势能面Fig.9 The potential energy surface of p-dibromobenzene molecule under different external electric fields |
采用密度泛函理论在B3LYP/6-311+G(d, p)基组水平上计算了不同外电场作用下对二溴苯分子的基态几何结构、 光谱特性和解离特性, 包括两个C—Br键长、 总能量、 偶极矩、 能级轨道分布、 能隙、 红外光谱、 紫外吸收光谱和两个键的势能曲线以及势能面, 分析了加外电场对这些性质的影响。 研究表明, 在外电场作用下, 对二溴苯分子的结构发生较大改变。 随着正向外电场的增加, 对二溴苯分子的3C—12Br键长逐渐减小, 6C—11Br键长逐渐增大; 随着正向外电场的增加, 分子体系总能量减小, 而偶极矩发生相反的变化; 对二溴苯分子的能隙逐渐减小。 对二溴苯分子的振动频率随外电场增强发生红移, C—Br键伸缩振动处的峰强度逐渐减小; 随着外电场的增强, 分子在处于紫外波段的紫外吸收光谱吸收峰峰值先略微增大, 后迅速降低, 峰值约减小一半, 峰形变宽。 对二溴苯分子C和Br原子间能量随外电场的增强逐渐降低, 当外电场强度为0.02 a.u.时, 分子的两个C—Br键逐步断裂, 发生逐步解离现象; 当外电场强度为0.02 a.u.时, 分子势能面的对角线降低, 出现另一个解离通道, 可能发生协同解离。 本研究计算得到了对二溴苯分子的基态几何数据和解离特性, 研究表明可对分子进行外电场降解、 收集, 且观察到了分子的协同解离现象, 这将为实验研究对二溴苯分子的协同解离提供理论指导。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|