

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于深度学习的冠状病毒刺突蛋白拉曼特征峰的理论研究
[倪爽 , 温家星, 周民杰, 黄景林, 乐玮, 陈果, 何智兵, 李波, 赵松楠, 赵宗清, 杜凯
, 温家星, 周民杰, 黄景林, 乐玮, 陈果, 何智兵, 李波, 赵松楠, 赵宗清, 杜凯* ]
, 温家星, 周民杰, 黄景林, 乐玮, 陈果, 何智兵, 李波, 赵松楠, 赵宗清, 杜凯]
|
|


作者简介: 倪 爽, 1984年生, 中国工程物理研究院助理研究员 e-mail: nishuang@163.com
持续一年的新冠疫情对全球的经济造成了巨大破坏, 为了有效控制新冠疫情, 快速检测新冠病毒(SARS-CoV-2)是一个急需解决的问题。 新冠病毒的刺突蛋白(spikeprotein)是拉曼光谱技术检测新冠病毒的检测点, 构建刺突蛋白拉曼特征峰模型对于发展拉曼检测技术快速检测新冠病毒具有重要作用。 基于简化的激子模型, 利用深度神经网络技术, 构建了刺突蛋白的酰胺Ⅰ、 Ⅲ特征峰模型, 并结合已知可以感染人类的七种冠状病毒(HCoV-229E, HCoV-HKU1, HCoV-NL63, HCoV-OC43, MERS-CoV, SARS-CoV和SARS-CoV-2)刺突蛋白的实验结构, 分析了七种冠状病毒刺突蛋白酰胺Ⅰ、 Ⅲ特征峰的区别。 计算结果表明, 七种冠状病毒可以根据毒刺突蛋白的酰胺Ⅰ、 Ⅲ特征峰划分为四个组: SARS-CoV-2, SARS-CoV, MERS-CoV形成一个组; HCoV-HKU1, HCoV-NL63形成一个组; HCoV-229E和HCoV-OC43各自独立形成一个组。 相同组的冠状病毒刺突蛋白酰胺Ⅰ、 Ⅲ峰频率较为接近, 通过酰胺Ⅰ、 Ⅲ峰的频率较难区分刺突蛋白; 不同组的冠状病毒刺突蛋白酰胺Ⅰ、 Ⅲ特征峰差异较大, 刺突蛋白可以通过拉曼技术区分开来。 该结果为发展拉曼检测技术快速检测新冠病毒提供了定性判断的理论依据。
, WEN Jia-xing, ZHOU Min-jie, HUANG Jing-lin, LE Wei, CHEN Guo, HE Zhi-bing, LI Bo, ZHAO Song-nan, ZHAO Zong-qing, DU Kai
COVID-19, which has lasted for a year, has caused great damage to the global economy. In order to control COVID-19 effectively, rapid detection of COVID-19 (SARS-CoV-2) is an urgent problem. Spike protein is the detection point of Raman spectroscopy to detect SARS-CoV-2. The construction of spike protein Raman characteristic peaks plays an important role in the rapid detection of SARS-CoV-2 using Raman technology. In this paper, we used Deep Neural Networks to construct the amideⅠ and Ⅲ characteristic peak model of spike proteins based on simplified exciton model, and combined with the experimental structures of seven coronaviruses (HCoV-229E, HCoV-HKU1, HCoV-NL63, HCoV-OC43, MERS-CoV, SARS-CoV, SARS-CoV-2) spike proteins, analyzed the differences of amideⅠ andⅢ characteristic peaks of seven coronaviruses. The results showed that seven coronaviruses could be divided into four groups according to the amideⅠ and Ⅲ characteristic peaks of spike proteins: SARS-CoV-2, SARS-CoV, MERS-CoV form a group; HCoV-HKU1, HCoV-NL63 form a group; HCoV-229E and HCoV-OC43 form a group independently. The frequency of amideⅠ and Ⅲ in the same group is relatively close,and it is difficult to distinguish spike proteins by the frequency of amideⅠ and Ⅲ; the characteristic peaks of amideⅠ and Ⅲ in different groups are quite different, and spike proteins can be distinguished by Raman spectroscopy. The results provide a theoretical basis for the development of Raman spectroscopy for rapid detection of SARS-CoV-2.
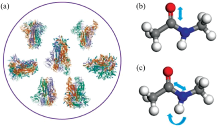
新冠疫情对全球的经济造成了巨大破坏。 为了有效控制新冠疫情, 快速检测新冠病毒(严重急性呼吸综合症冠状病毒2, severe acute respiratory syndrome coronavirus 2, SARS-CoV-2)是一个急需解决的问题。 新冠病毒的刺突蛋白是病毒攻击人体的钥匙, 且在病毒表面大量分布, 因此成为拉曼光谱技术检测新冠病毒的检测点[1, 2, 3]。 要实现刺突蛋白的拉曼技术检测, 关键之一在于构建刺突蛋白的拉曼特征峰。 鉴于纯刺突蛋白的拉曼谱图较难获得且成本较高, 需要从理论上快速构建刺突蛋白拉曼特征峰。 此外, 人类已知可以感染的七种冠状病毒刺突蛋白的结构[4, 5, 6, 7, 8, 9]相近[图1(a)], 是否可以通过拉曼技术区分他们对于新冠病毒的准确检测是一个十分重要的问题。 基于理论构建的刺突蛋白拉曼特征峰, 可以分析七种冠状病毒刺突蛋白拉曼特征峰的不同, 为实验提供指导。
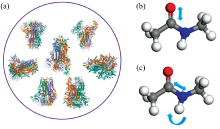 | 图1 (a)七种冠状病毒刺突蛋白的结构示意图(蛋白结构图取自https: //www.rcsb.org/); (b), (c)N-甲基乙酰胺中酰胺Ⅰ 、 Ⅲ 峰的振动模式示意图(b为酰胺Ⅰ 振动模式; c为酰胺Ⅲ 振动模式)Fig.1 (a) Structure of spike proteins of seven coronaviruses (The protein structure diagram is taken from https://www.rcsb.org/); (b), (c) Vibration mode diagram of amide Ⅰ and Ⅲ peaks in N-methylacetamide (b is amide Ⅰ vibration mode; c is amide Ⅲ vibration mode) |
拉曼检测主要是测量分子振动模式的频率和强度, 理论上可以通过密度泛函理论(DFT)计算[10, 11]。 然而, 刺突蛋白有数万个原子, 较难直接使用DFT方法计算。 蛋白骨架是蛋白的特征结构, 骨架结构中的酰胺单元振动形成的酰胺Ⅰ (主要是C=O伸缩振动, 还有一些贡献来自于C— N伸缩)、 Ⅲ 峰(主要贡献来自于N— H弯曲和C— N伸缩振动的组合, 还有一些贡献来自于C=O平面内弯曲和C— C伸缩振动)[图1(b, c)]是蛋白的特征拉曼谱峰。 理论上基于激子模型可以构建蛋白的酰胺Ⅰ 峰[12, 13, 14]。 激子模型假设体系的简正模可以用局域模来表示, 激子模型哈密顿量可以用局域模态|i> 来表示
式(1)中: ε i为局域模频率, β ij为局域模之间的相互作用, 激子模型哈密顿量可以写成矩阵形式
式(2)中: n为局域模的数量, 对角化H矩阵可以获得激子的频率及其对应特征矢量(局域模展开为简正模的线性展开系数)。 对于蛋白酰胺Ⅰ 峰而言, 其局域模近似为组成蛋白骨架的每个酰胺单元的酰胺Ⅰ 振动模。
本文关注的重点是定性分析七种冠状病毒刺突蛋白拉曼特征峰的差异而非绝对值且在激子模型中H矩阵对角元的值远大于非对角元的值。 因此, 本文采用简化的激子模型, 仅考虑结构变化对H矩阵对角元的校正而将非对角元舍去。
为了校正对角元, 需要对七种刺突蛋白结构中的每个酰胺单元计算拉曼谱图, 每个刺突蛋白有数千个酰胺单元, 逐个计算拉曼谱图费时且不利于统一误差。 江俊[15, 16]等近期提出了基于机器学习构建分子结构和拉曼性质(频率、 强度)之间映射关系的策略。 其先通过分子动力学构建分子的动力学结构, 然后计算结构的拉曼频率以及强度, 最后使用机器学习拟合结构和性质的映射关系。 本文基于此策略, 构建酰胺单元结构和拉曼峰频率以及强度之间的模型。
文献中多数研究集中在酰胺Ⅰ 峰, 对酰胺Ⅲ 拉曼峰研究较少。 本文基于深度学习技术, 从理论上构建蛋白酰胺单元结构和酰胺Ⅰ 、 Ⅲ 拉曼特征峰的映射关系, 然后统计七种冠状病毒刺突蛋白的结构差异, 带入模型中获得拉曼特征峰。 最后通过洛伦兹线型展开谱线获得谱图, 并比较谱图的差异。
使用N-甲基乙酰胺(NMA)分子来模拟蛋白的酰胺单元[图1(b, c)], 构建酰胺Ⅰ 、 Ⅲ 峰的深度学习模型。 首先, 通过VASP软件, 采用分子动力学的方法构建10 000个NMA分子随时间演化的结构, 未考虑H2O的影响。 然后通过Gaussian09软件计算这些结构对应的拉曼谱峰, 最后通过深度学习技术构建NMA分子结构特征和酰胺Ⅰ 、 Ⅲ 峰频率以及强度的映射关系。 VASP计算采用GGA-PBE泛函, 周期性的边界条件为14.6× 14.6× 14.6 Å 3, 平面波的截断能设置为400 eV, 能量收敛标准为1× 10-5 eV, 分子动力学模拟选择正则系综(NVT), 时间步长1 fs, 温度为300 K, 总共模拟10 000步。 Gaussian09计算拉曼谱峰基于B3LYP杂化泛函水平, 基组使用6-311++G(d, p)。
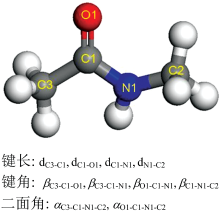
深度学习模型由1个输入层, 3个隐藏层以及1个输出层组成, 对于每一个隐藏层, 使用线性修正单元(Rectified Linear Unit)激活函数。 对于NMA分子, 选用10个结构特征来描述(4个键长, 4个键角, 2个二面角, 如图2)。 为了增强模型的鲁棒性, 对输入特征以及输出结果进行了归一化处理。
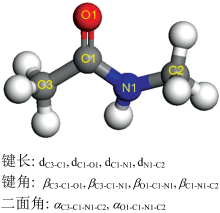 | 图2 用来预测NMA分子拉曼位移以及强度的描述符Fig.2 The descriptors of NMA molecular used to predict Raman shifts and intensities |
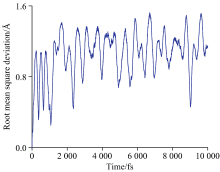
基于分子动力学构建了10 000个NMA分子结构, 计算了其均方根误差, 为1.06 Å (图3), 这说明通过动力学演化得到的NMA分子结构变化较大且相关性较小。
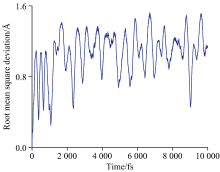 | 图3 NMA分子结构的均方根误差Fig.3 Root mean square deviation of NMA molecular structures |
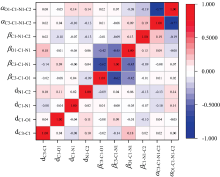
同时, 计算了NMA分子特征间的皮尔逊相关系数(Person correlation coefficient), 皮尔逊相关系数度量两个变量之间的相关程度, 从图4中可以看出, 除了二面角α C3-C1-N1-C2和α O1-C1-N1-C2之间的相关系数较大以外, 其余的系数基本都接近于0, 这说明NMA分子所选特征相关性很小, 二面角α C3-C1-N1-C2和α O1-C1-N1-C2之间的相关性主要源于NMA中N1上的孤对电子和羰基(C1=O1)具有明显的共轭作用, 使得C3, O1, C1, N1和C2原子基本共面所致。
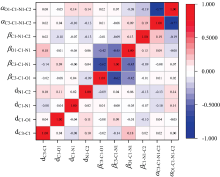 | 图4 NMA描述符间的皮尔逊相关系数Fig.4 Pearson correlation coefficient between NMA molecular descriptors |
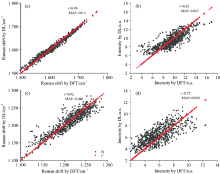
酰胺Ⅰ 、 Ⅲ 峰的拉曼位移(振动频率)和拉曼强度是深度学习模型的目标, 所有的数据分成两部分, 训练集和测试集。 测试集取700个数据, 剩余的为训练集数据。 通过训练集训练出模型以后, 在测试集上评估模型的泛化能力。 酰胺Ⅰ , Ⅲ 峰的振动频率, 拉曼强度通过深度学习模型计算的结果和DFT计算的结果比较如图5所示。 对于酰胺Ⅰ 的振动频率模型[图5(a)], 皮尔逊相关系数(r)为0.99, 表明酰胺Ⅰ 频率模型有很高的预测精度; 对于酰胺Ⅲ 的振动频率模型[图5(c)], r为0.92, 表明该模型也有很高的精度, 略逊于酰胺Ⅰ 的振动频率模型。 这可能有两点原因构成, 一是酰胺Ⅰ 的振动模式相对简单, C=O伸缩振动占据很高的比例, 相比之下酰胺Ⅲ 的振动模式复杂一些, 除了C— N伸缩、 N— H弯曲振动外, 一些键长、 键角的振动也占一定的比例; 二是酰胺Ⅰ 的振动频率所对应的能量空间相对局域, 酰胺Ⅰ 的振动和其他振动模式之间的耦合较小, 更适合激子模型。 对于酰胺Ⅰ 的拉曼强度模型[图5(b)], r为0.81, 较为良好却不够精确。 对于酰胺Ⅲ 的拉曼强度模型[图5(d)], r只有0.72, 精度相对略差。 拉曼强度模型精度低于振动频率模型的原因可能是拉曼强度公式为能量的三阶偏导, 其公式的复杂程度远超过频率(体系的能量), 因此学习效果略差。
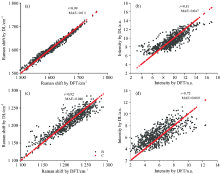 | 图5 酰胺Ⅰ 峰的拉曼位移(a)、 强度 (b)以及酰胺Ⅲ 峰的拉曼位移(c)、 强度(d)通过深度学习模型和DFT计算的结果比较2.2 七种冠状病毒刺突蛋白骨架的结构特征Fig.5 The Raman shift (a) and intensity (b) of amide I peak and the Raman shift (c) and intensity (d) of amide Ⅲ peak were compared by deep learning model and DFT calculation |
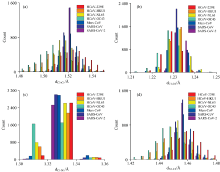
基于实验上的冠状病毒刺突蛋白结构[4, 5, 6, 7, 8, 9](PDB code: 6u7h, 5i08, 5szs, 6ohw, 5x59, 5x58, 6vsb), 统计其骨架特征键长、 键角、 二面角的分布。
从图6中可以看出, C1— N1和C1— O1的键长分布较为集中, 这主要是N1的孤对电子和(C1=O1)发生共轭导致C1— N1和C1— O1均有双键的性质, 键的力常数较大, 不易改变。 HCoV-OC43刺突蛋白骨架的键长相比于其他病毒蛋刺突蛋白结构显著偏低, 这会导致和键长相关的酰胺Ⅰ 、 Ⅲ 峰的频率偏高。 HCoV-229E的C1— O1键长和HCoV-OC43的相近[图6(b)], 由于C1— O1键长是酰胺Ⅰ 峰频率的决定性因素, 因此HCoV-229E和HCoV-OC43刺突蛋白的酰胺Ⅰ 峰相近。 HCoV-HKU1, HCoV-NL63刺突蛋白骨架的键长大于HCoV-OC43的键长但小于MERS-CoV, SARS-CoV, SARS-CoV-2的键长, 因此, HCoV-HKU1, HCoV-NL63刺突蛋白的酰胺Ⅰ 、 Ⅲ 峰的频率低于HCoV-OC43但高于MERS-CoV, SARS-CoV, SARS-CoV-2。
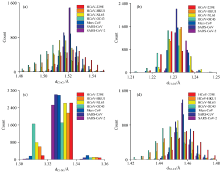 | 图6 七种冠状病毒刺突蛋白骨架的键长特征统计分布图Fig.6 Statistical distribution of bond length of seven coronaviruses spike protein backbone |
从键角[图7(a— d)]的分布可以看出: 键角的分布较为广泛且均匀, 说明键角变化的力常数较小, 七种冠状病毒的键角变化相差不大, 键角对酰胺特征峰的影响较小。
 | 图7 七种冠状病毒刺突蛋白骨架的键角和二面角特征统计分布图Fig.7 Statistical distribution of bond angle anddihedral angle of seven coronaviruses spike protein backbone |
从二面角的分布[图7(e, f)]中可以看出, 七种冠状病毒刺突蛋白的二面角均在180° (3.14弧度)附近, 这是由于酰胺平面共轭导致的。
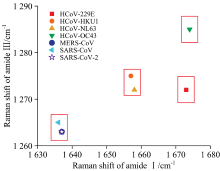
根据前面获得的酰胺Ⅰ 、 Ⅲ 峰的模型(酰胺单元的10个特征和酰胺Ⅰ 、 Ⅲ 振动峰的频率以及强度的映射关系), 从七种冠状病毒刺突蛋白的实验结构中计算出每个酰胺单元的10个特征, 带入到模型中获得每个酰胺单元的酰胺Ⅰ 、 Ⅲ 拉曼峰(振动频率和强度), 最后通过洛伦兹线型将每个酰胺单元的Ⅰ 、 Ⅲ 拉曼峰展开获得七种冠状病毒刺突蛋白的酰胺Ⅰ 、 Ⅲ 谱带(图8)。 从图中可以看出, 七种冠状病毒刺突蛋白的酰胺Ⅰ 、 Ⅲ 谱带根据最高峰频率可以各分成三个组。 对于酰胺Ⅰ 峰, SARS-CoV-2, SARS-CoV, MERS-CoV刺突蛋白频率相近, 其频率值在1 636~1 637 cm-1区间; HCoV-HKU1, HCoV-NL63刺突蛋白频率相近, 其频率值在1 657~1 658 cm-1区间; HCoV-229E, HCoV-OC43刺突蛋白频率相近, 其频率值在1 673~1 674 cm-1区间。 对于酰胺Ⅲ 峰, SARS-CoV-2, SARS-CoV, MERS-CoV刺突蛋白频率相近, 其频率值在1 263~1 265 cm-1; HCoV-229E, HCoV-HKU1, HCoV-NL63刺突蛋白频率相近, 其频率值在1 272~1 275 cm-1; HCoV-OC43刺突蛋白单独一个频率, 其频率值为1 285 cm-1。
 | 图8 七种冠状病毒刺突蛋白的酰胺Ⅰ 、 Ⅲ 拉曼特征峰比较 (a): 酰胺Ⅰ ; (b): 酰胺Ⅲ Fig.8 Comparison of Raman characteristic peaks of amide Ⅰ and Ⅲ of seven coronavirus spike proteins (a): Amide Ⅰ ; (b): Amide Ⅲ |
根据上面的分析, 可以根据酰胺Ⅰ 、 Ⅲ 峰的频率划分七种冠状病毒, 如图9所示, 七种冠状病毒分为四组: SARS-CoV-2, SARS-CoV, MERS-CoV在同一个组; HCoV-HKU1, HCoV-NL63为一组; HCoV-229E一组; HCoV-OC43一组。 不同组之间其刺突蛋白特征峰的数值差异较大, 可以区分开来。 同组中的刺突蛋白特征峰的数值差异较小, 较难区分。
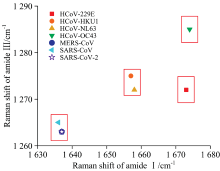 | 图9 通过酰胺Ⅰ 、 Ⅲ 特征峰频率区分七种冠状病毒Fig.9 Seven coronaviruses were distinguished by the frequency of amide Ⅰ and Ⅲ characteristic peaks |
基于深度学习的技术构建了刺突蛋白酰胺Ⅰ 、 Ⅲ 特征拉曼峰模型, 结合实验上的冠状病毒刺突蛋白结构, 获得了七种冠状病毒刺突蛋白的酰胺Ⅰ 、 Ⅲ 拉曼特征峰, 通过比较七种冠状病毒刺突蛋白的拉曼特征峰, 可以把七种冠状病毒分为四组: SARS-CoV-2, SARS-CoV, MERS-CoV一组; HCoV-HKU1, HCoV-NL63一组; HCoV-229E一组; HCoV-OC43一组。 不同组的冠状病毒特征峰差异较大, 可以区分开来; 同一组的冠状病毒特征峰差异较小, 较难区分。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|