





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
利巴韦林、 磷酸氯喹、 盐酸阿比多尔等抗病毒药物的红外光谱表征
[陈靖奕1  , 朱南
, 朱南2 , 昝佳男3 , 肖子康1 , 郑晶1 , 刘畅1 , 沈睿1 , 王芳1, 3, * , 刘云飞3 , 蒋玲3 ]
, 朱南, 昝佳男, 刘云飞|
|


作者简介: 陈靖奕, 2000年生, 南京晓庄学院电子工程学院本科生 e-mail: 1830419583@qq.com;朱 南, 1986年生, 南京市产品质量监督检验院工程师 e-mail: nzhuwork@126.com;陈靖奕, 朱 南: 并列第一作者
自新冠肺炎(COVID-19)疫情爆发以来, 国内外多家研究机构和企业都在加快推进新冠病毒(SARS-CoV-2)抗体药物的研发。 药物多晶型限制了有效药物的研发进度。 药物生产、 存储和使用环境影响了药物的稳定性。 红外光谱作为一种快速无损检测手段, 可从振动光谱反映出药物结构、 晶型甚至生产工艺上的差异大大提高了研发效率。 首次以三种临床试验被认为治疗新冠肺炎有效药物: 磷酸氯喹, 利巴韦林和盐酸阿比多尔为例, 利用傅里叶红外光谱仪测试得到它们在远红外(1~10 THz)和中红外(400~4 000 cm-1)波段的振动光谱。 远红外光谱中, 利巴韦林的特征峰位于: 2.01, 2.68, 3.37, 4.05, 4.83, 5.45, 5.92, 6.42和7.14 THz附近; 磷酸氯喹的特征峰位于: 1.26, 1.87, 2.37, 3.06, 3.78, 5.09和6.06 THz附近; 盐酸阿比多尔的特征峰位于: 2.24, 3.14, 3.72, 4.25和5.38 THz附近。 结合密度函理论, 选择B3LYP杂化泛函和6-311++G(d,p)基组, 利用Crystal14和Gaussian16软件分析了光谱中所有特征峰对应的振动模式, 实现了对振动光谱的精确指认。 远红外波段, 振动模式源自分子的集体振动。 中红外波段, 2 800 cm-1以下, 振动模式主要源自基团的面内外弯曲和摇摆; 2 800 cm-1以上, 振动模式过渡为C—H, O—H和N—H键的面内伸缩。 以考虑了周期性边界条件的晶体结构作为理论计算的初始构型, 会让理论光谱与实验光谱更加吻合, 尤其是在远红外波段和中红外400~1 000 cm-1的低频段。 该研究对深入理解磷酸氯喹, 利巴韦林和盐酸阿比多尔等抗病毒药物的药学特性, 药物间相互作用, 控制药物生产过程, 指导药物存储和使用有重大意义。
, ZHU Nan, ZAN Jia-nan, LIU Yun-feiCHEN Jing-yi and ZHU Nan: joint first authors
Since the outbreak of novel coronavirus pneumonia (COVID-19), many research institutes and enterprises at home and abroad have been accelerating the research of COVID-19 (SARS-CoV-2) antibody drugs. However, the research on effective drugs was limited by the drug polymorphisms. The environment of drug production, storage and use also affected the stability of the drug. As a fast, non-destructive testing method, infrared spectroscopy can reflect the differences in drug structure, crystal form and even manufacturing technique to the vibration spectrum, which greatly improves the efficiency of R&D (research and development). In this paper, three clinical trials were considered effective drugs for the treatment of COVID-19: Chloroquine diphosphate, Ribavirin and Abidol hydrochloride. Their far-infrared spectrum (1~10 THz) and mid-infrared spectrum (400~4 000 cm-1) were measured by Fourier transform infrared spectrometer (FTIR). In the far-infrared spectrum, the characteristic peaks of Ribavirin were around 2.01, 2.68, 3.37, 4.05, 4.83, 5.45, 5.92, 6.42 and 7.14 THz; the characteristic peaks of Chloroquine phosphate were near 1.26, 1.87, 2.37, 3.06, 3.78, 5.09 and 6.06 THz; the characteristic peaks of Abidol hydrochloride were located near 2.24, 3.14, 3.72, 4.25 and 5.38 THz. Based on density functional theory, the B3LYP hybrid functional and 6-311++G (d, p) basis sets were selected to analyze the vibrational modes corresponding to all characteristic peaks in the spectrum using Crystal14 and Gaussian 16 software, and the accurate identification of the vibration spectrum was realized. The vibrational modes originated from the molecules’ collective vibration in the far infrared region. In the mid-infrared band, below 2 800 cm-1, the vibrational modes mainly came from the in-plane and out-of-plane bending and rocking of the group; Above 2 800 cm-1, the vibrational modes transited to the in-plane stretching of C-H, O-H and N-H bonds. Taking the crystal structure with periodic boundary conditions as the initial configuration of the theoretical calculation would make the calculated spectrum more consistent with the experimental one, especially in the far-infrared band and the low-frequency band of mid-infrared (400~1 000 cm-1). This study was of great significance to deeply understand the pharmaceutical characteristics, drug interactions, control of drug production process, and guide the storage and use of antiviral drugs such as Chloroquine phosphate, Ribavirin and Abidol hydrochloride.
磷酸氯喹(chloroquine phosphate), 利巴韦林(ribavirin)和盐酸阿比多尔(arbidol hydrochloride)为经过临床试验被认为治疗新冠肺炎有效的临床药物[1, 2], 均被列入《新型冠状病毒肺炎诊疗方案》中。 自新冠肺炎(COVID-19)疫情发生以来, 国内外多家研究机构和企业正在加快推进新冠病毒(SARS-CoV-2)抗体药物的研发。 药物多晶型与药物的熔点、 稳定性、 溶解性、 密度等物理化学性质密切相关, 从而影响药效及药物间的相互作用[3]。 在制药工艺中, 将这些性质与加工特性、 储存特性结合在一起考虑是非常必要的。 研究发现, 利巴韦林作为临床一线抗病毒治疗药物, 用于病毒引起的肺炎、 支气管炎、 皮肤疱疹等疾病的治疗。 其存在晶A、 晶B、 晶C、 和晶D四种晶型[4]。 药动学实验表明, 只有晶A型在临床治疗中有显著优势。 远红外光谱技术(以下称之为: 太赫兹光谱技术)作为一种快速检测手段, 可表征分子间的弱相互作用, 如氢键、 范德华力、 晶格间的堆积力等。 多项研究表明[5], 利用太赫兹光谱技术可以快速筛选出优势晶型药物, 大大地缩短了研发时间, 降低了研发成本。
高温、 高湿以及暴露时间均是影响药物稳定性的关键因素, 轻则影响治疗效果, 重则关乎生命安全。 传统的药物稳定性检测手段包括质谱分析法、 拉曼光谱分析法、 红外光谱分析法(特指中红外)、 热重分析法、 X射线粉末衍射分析法以及扫描电子显微分析法等在判别药物结构及其稳定性时均存在一定的弊端, 不能同时将结构、 晶型、 甚至生产工艺给药效带来的影响反映到图谱的变化上。
通过测试和分析三种临床试验认为治疗新冠肺炎有效药物: 磷酸氯喹, 利巴韦林和盐酸阿比多尔的远红外(以下称之为太赫兹)和中红外(以下称之为红外)光谱, 获取每种药物光谱中所有特征峰位置, 并通过理论分析得到其对应的振动模式。 不仅可以有效地检测药物结构是否发生改变, 发生了什么改变, 还可以实时监测优势药物晶型是否在向其他晶型转变。 该研究对深入理解磷酸氯喹, 利巴韦林和盐酸阿比多尔等抗病毒药物的药学特性, 药物间相互作用, 控制药物生产过程, 指导药物存储和使用有重大意义。
采用德国布鲁克公司的VERTEX 80V型傅里叶红外光谱仪, 频谱范围为: 10~15 500 cm-1, 通过不同的光学选件, 来覆盖远红外、 中红外和近红外光谱区域。 中红外波段有效频率范围为: 400~4 000 cm-1; 远红外波段配置4个分束器: 6, 25, 50和125 μ m, 本工作选择的是6 μ m分束器, 有效频率范围为: 30~680 cm-1(0.9~20.4 THz); 真空环境下的分辨率可达2 cm-1; 信噪比优于50 000∶ 1(峰-峰值)。 中红外光谱测试时, 光学腔始终保持在真空环境, 样品腔处在标准大气压下; 远红外光谱测试时, 通过给样品腔和光学腔抽真空的方式减少环境中水汽对光谱的影响。 所有测试均在室温下完成, 测试原理和方法已经在前期研究中详细阐述, 在这里不再赘述[6]。
三种抗病毒药物磷酸氯喹(C18H26ClN3· 2H3PO4), 利巴韦林(C8H12N4O5)和盐酸阿比多尔(C22H25BrN2O3S· HCl)购于Sigma-Aldrich公司, 纯度99%以上, 样品测试前均未经进一步纯化。 由于样品在远红外波段的吸收很强, 光谱测试前, 将纯样与聚乙烯粉末以1∶ 10的质量比混合, 研磨均匀后压成0.5~1 mm厚, 直径为13 mm的薄片。 同时制备纯聚乙烯样品薄片作为测试背景, 纯聚乙烯样品的质量需与测试样品中聚乙烯含量(质量)相同, 这样可以排除聚乙烯吸收的影响。 中红外光谱测试前样品制备相对简单, 只需将一定质量的样品跟溴化钾混合, 充分研磨后压成直径为13 mm的透明薄片即可。
以利巴韦林为例, 计算了其基于晶体结构的振动光谱; 初始晶体结构来自剑桥晶体库, 对应的refcode为VIRAZL[7], 正交(斜方)晶型, 晶格参数为: a=14.863, b=7.512, c=8.788, α =90.000, β =90.000, γ =90.000。 其单分子和单晶胞结构如图1所示, 每个晶胞包含4个单分子。 利用Crystal14软件计算得到优化后结构的振动光谱, 计算结果无虚频, 选择B3LYP杂化泛函和6-311++G(d, p)基组, 同时添加了GRIMME伦敦色散校正系数, TOLINTEG=(10 10 10 10 20)。
 | 图1 利巴韦林单分子(优化后, a)和晶体(b)结构Fig.1 The monomer (after optimization, a) and crystal structure (b) of Ribavirin |
因磷酸氯喹和盐酸阿比多尔分子量较大, 需要耗费较多的计算资源, 且剑桥晶体库中没有合适的晶体结构, 故只计算了其优化后单分子结构的振动光谱。 优化前和优化后的磷酸氯喹和盐酸阿比多尔单分子结构见图S1, 磷酸氯喹经优化后磷酸分子中O— H键均被拉长, 其中O50— H53键拉长后发生断裂, 使得N16— H53之间形成了氢键。 利用Guassian16软件, 结合密度泛函理论(density functional theory, DFT), 选择B3LYP杂化泛函和6-311++G(d, p)基组进行理论振动光谱分析, 计算结果无虚频, 拟合半宽高均为4 cm-1。
 | 图S1 优化前(a)和优化后(b)的盐酸阿比多尔和磷酸氯喹单分子结构Fig.S1 The structures of Arbidol hydrochloride and Chloroquine phosphate before (a) and after (b) optimization |
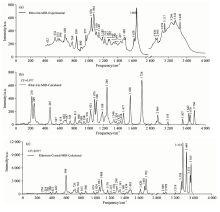
盐酸阿比多尔、 利巴韦林和磷酸氯喹等三种抗病毒药物的太赫兹理论(基于单分子结构)和实验光谱, 如图2所示。 因理论计算初始结构基于单分子, 只考虑了分子内的相互作用力; 而实验光谱基于无定形固态晶体结构, 除了分子内的相互作用力外, 还存在分子间的相互作用力, 理论研究表明这些分子间的弱相互作用力(如晶胞堆积力、 氢键和范德华力等)均落在太赫兹波段[8], 这就造成了理论和实验光谱之间存在一定的偏差。 从图2(b)可以看出, 盐酸阿比多尔特征峰主要位于: 2.24, 3.14, 3.72, 4.25和5.38 THz附近; 利巴韦林特征峰主要位于: 2.01, 2.68, 3.37, 4.05, 4.83, 5.45, 5.92, 6.42和7.14 THz附近; 磷酸氯喹特征峰主要位于: 1.26, 1.87, 2.37, 3.06, 3.78, 5.09和6.06 THz附近。 盐酸阿比多尔主要振动模式源于HCl和H2O分子的面外摇摆, 除此之外还伴随着苯硫甲基、 吲哚、 二甲氨甲基和羟基的振转动; 利巴韦林的主要振动模式来自集体振动以及呋喃核糖基和羧酰胺的面外摇摆; 磷酸氯喹的主要振动模式源自磷酸分子和二乙基的振转动。
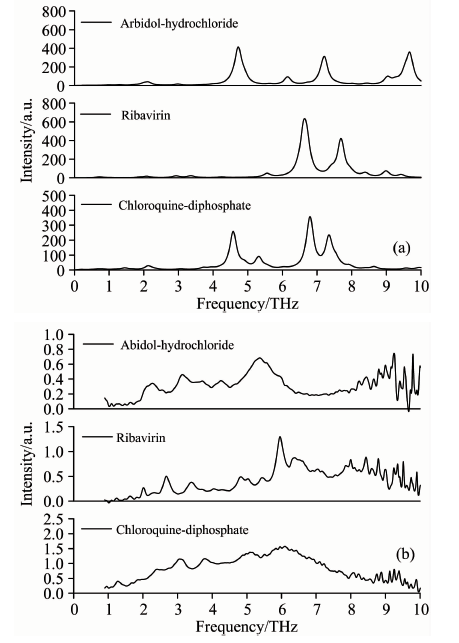 | 图2 三种抗病毒药物的理论(a, 基于单体)和 实验光谱(b)对比Fig.2 Comparison of theoretical (a, based on the monomer) and experimental (b) spectra of three antiviral drugs |
前期, 我们对构成DNA/RNA的碱基、 碱基对、 特征片段的太赫兹光谱详细研究[9, 10, 11, 12, 13]后发现, 太赫兹光谱反映的是分子内和分子间的一种集体振动, 无论是单体、 二聚体还是多聚体的理论光谱均不能很好的重现实验结果。 只有将考虑了周期性边界条件的晶体结构作为理论计算的初始构型, 才能让理论和实验结果一致吻合。 图3为利巴韦林理论(基于晶体)和实验光谱对比图, 从图中可以发现, 除部分特征峰发生了简并外, 理论与实验光谱较为吻合, 说明考虑了周期性边界条件的密度泛函理论可以较好地描述实验结果。
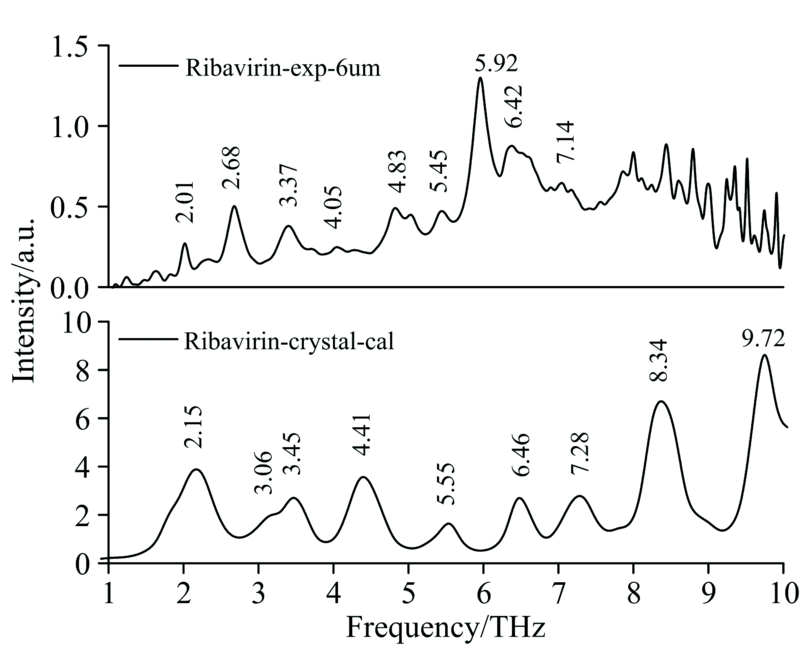 | 图3 利巴韦林太赫兹实验和理论(基于晶体)光谱对比Fig.3 Comparison of experimental and calculation (based on the crystal) THz spectra of Ribavirin |
表1给出了利巴韦林实验和理论(基于晶体)太赫兹光谱中所有特征峰位置及其对应的振动模式, 从表中可以看出, 10 THz以下的振动模式均表现为晶胞中所有分子参与的集体振动, 以三氮唑的面外弯曲、 羧酰胺的面外摆动和呋喃核糖基的弱面内伸缩为主。
| 表1 利巴韦林实验和理论(基于晶体)太赫兹 光谱特征峰位置及其对应的振动模式 Table 1 Experimental and calculated (based on the crystal) THz vibration frequencies and corresponding vibrational modes of Ribavirin |
太赫兹光谱主要源于分子的集体振动, 分子结构和晶型的改变、 甚至生产工艺上的差异均能使其光谱发生变化, 但也因其振动模式过于复杂, 难以精准解析。 中红外光谱主要源自部分基团或者原子的振动, 虽只能将结构的变化反映到光谱的变化上, 但因其振动模式简单, 可对光谱精准解析。 将太赫兹光谱和中红外光谱相结合, 既可以从结构、 晶型、 生产工艺上发现分子间的差异, 还可对光谱进行比较深入的解析。
图4为盐酸阿比多尔实验和理论(基于单体)中红外光谱图, 有效频率范围为400~4 000 cm-1, 因1 800~2 800 cm-1范围内无特征, 故略去。 从图中可以看出, 理论和实验光谱较为吻合。 因理论计算基于谐振子模型, 而实验环境复杂, 一般会通过给理论光谱乘上一个频率校正因子, 来消除理论偏差, 校正因子的大小取决于选择的理论计算方法和基组。 文中选择基于密度泛函理论(DFT)的B3LYP杂化泛函和6-311++G(d, p)基组, 故乘上了0.977的频率校正因子[14]。
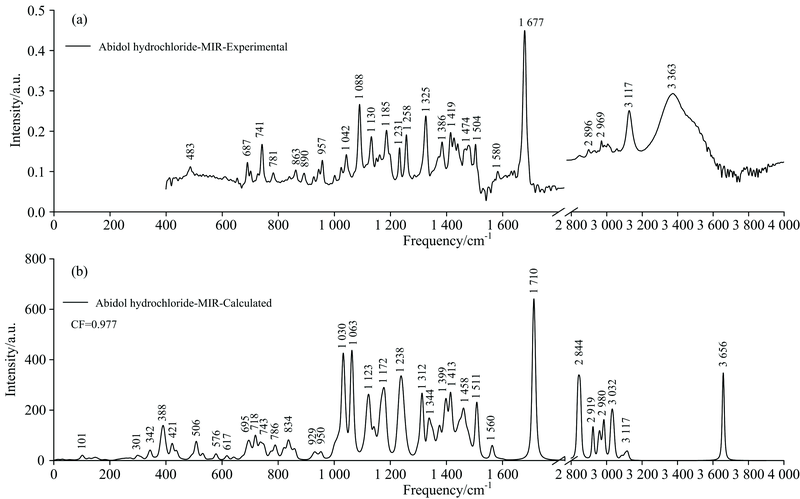 | 图4 盐酸阿比多尔实验(a)和理论(b, 基于单体)中红外光谱对比图, 频率范围400~4 000 cm-1Fig.4 Comparison of the experimental (a) and calculated (b, based on the monomer) MIR spectra of Abidol hydrochloride ranging between 400 and 4 000 cm-1 |
表2列出了盐酸阿比多尔在400~4 000 cm-1范围内中红外光谱中所有特征峰位置及其对应的主要振动模式。 从表中可以看出, 盐酸分子的振动峰在483 cm-1附近; 苯硫甲基的振动峰主要在687, 741和3 117 cm-1附近; 吲哚、 二甲氨甲基、 羧酸乙酯和甲基的振动贯穿整个中红外波段。 2 800 cm-1以下, 基团的振动以面内外弯曲和摇摆为主; 2 800 cm-1以上, 基团的振动过渡为面内伸缩。
| 表2 盐酸阿比多尔实验和理论(基于单体)中红外光谱特征峰位置及其对应的振动模式 Table 2 Experimental and calculated (based on the monomer) MIR vibration frequencies and corresponding vibrational modes for Abidol hydrochloride |
图5为磷酸氯喹实验和理论(基于单体)中红外光谱图, 有效频率范围为400~4 000 cm-1, 因1 800~2 400 cm-1范围内无特征, 故略去。 从图中可以看出, 理论和实验光谱较为吻合。 为了消除理论计算带来的偏差, 同样乘上0.977的频率校正因子[14]。
 | 图5 磷酸氯喹实验(a)和理论(b, 基于单体)中红外光谱对比图, 频率范围400~4 000 cm-1Fig.5 Comparison of the experimental (a) and calculated (b, based on the monomer) MIR spectra of Chloroquine phosphate ranging between 400 and 4 000 cm-1 |
表S1列出了磷酸氯喹在400~4 000 cm-1范围内中红外光谱中所有特征峰位置及其对应的主要振动模式。 从表中可以看出, 磷酸的振动峰主要集中在500~1 400 cm-1范围内, 呈现面内外弯曲和摇摆; 1 400~1 600 cm-1范围内, 以喹啉基、 二乙基和戊二胺的面内摇摆和弯曲为主; 2 600 cm-1以上, 基团的振动过渡为N— H, O— H和 C— H键面内伸缩。
| 表S1 磷酸氯喹实验和理论(基于单体)中红外光谱特征峰位置及其对应的振动模式 Table S1 Experimental and calculated (based on the monomer) MIR vibration frequencies and corresponding vibrational modes for Chloroquine phosphate |
图6为利巴韦林实验(与《药品红外光谱集第一卷》结果吻合, 光谱号22)和理论(基于单体和晶体)中红外光谱图, 有效频率范围为400~4 000 cm-1, 因1 800~2 800 cm-1范围内无特征, 故略去。 为了消除理论计算带来的偏差, 也乘上0.977的频率校正因子[14]。 与前面不同的是, 用于理论计算的初始结构除了单体外, 还对比了考虑周期性边界条件的晶体。 我们发现, 考虑了周期性边界条件的晶体结构的理论光谱与实验光谱吻合得更好, 尤其是400~1 000 c
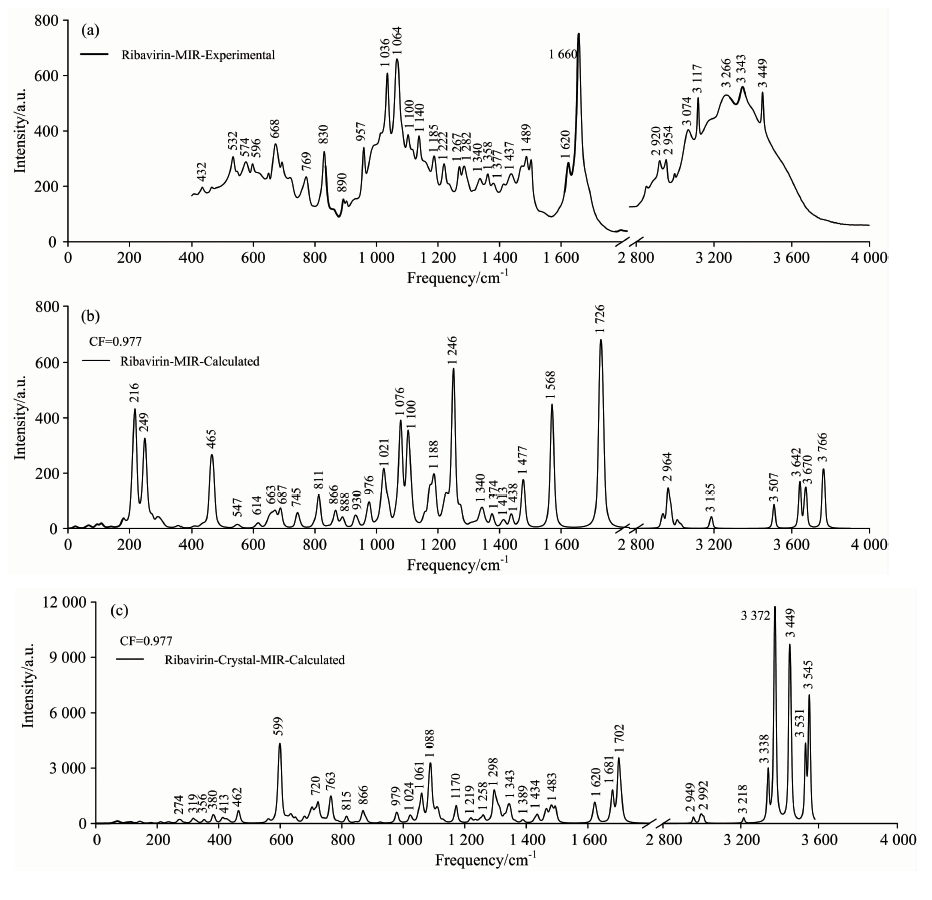 | 图6 利巴韦林实验(a)和理论(b, 基于单体; c, 基于晶体)中红外光谱对比图, 频率范围400~4000 cm-1Fig.6 Comparison of the experimental (a) and calculated (b, based on the monomer and (c) based on the crystal) MIR spectra of Ribavirin ranging between 400 and 4 000 cm-1 |
表S2列出了利巴韦林在400~4 000 cm-1范围内中红外光谱中所有特征峰位置及其对应的主要振动模式。 从表中可以看出, 1 000 cm-1以内既存在分子内作用力, 也存在一些分子间作用力, 如: 432 cm-1对应的振动模式中, 呋喃核糖上O— H键面内摇摆属于分子内作用, 羧酰胺上NH2键面外摇摆来自分子间作用。 1 000 cm-1以下的振动模式主要源自呋喃核糖基、 三氮唑和羧酰胺的面内外弯曲和摇摆, 1 000 cm-1以上的振动模式主要来自C— H, O— H, N— H和NH2键的面内摇摆和伸缩。 同样, 2 800 cm-1以上, 基团的振动过渡为面内伸缩。
| 表S2 利巴韦林实验和理论(基于单体* 和晶体#)中红外光谱特征峰位置及其对应的振动模式 Table S2 Experimental and calculated (based on the monomer* and the crystal#) MIR vibration frequencies and corresponding vibrational modes for Ribavirin |
以磷酸氯喹, 利巴韦林和盐酸阿比多尔这三种广谱抗病毒药物为例, 研究了它们的太赫兹和中红外振动光谱, 获得了光谱中所有特征峰信息。 既可以从结构、 晶型、 生产工艺上发现分子间的差异, 还可以对光谱进行比较深入的解析。
药物多晶是药物研发的壁垒之一, 生产、 存储和使用环境也是影响药物稳定性的关键因素。 红外光谱技术可同时将结构和晶型的变化反映到红外图谱上。 我们前期研究一种治疗高磷酸盐血症药物, 四水合碳酸镧时还发现, 三种不同工艺获得的药物, 拥有同样的PXRD图谱, 但药效却相差甚远, 利用太赫兹光谱可快速作出鉴别(与南京卡文迪许生物工程技术有限公司合作, 已申请发明专利), 筛选出最优药物[15]。
以考虑了周期性边界条件的利巴韦林晶体结构为例, 结合密度泛函理论和Crystal14软件, 选择B3LYP杂化泛函和6-311++G(d, p)基组, 考虑伦敦色散校正系数, 对其太赫兹和中红外光谱进行了表征。 研究表明, 理论和实验光谱一致吻合, 太赫兹波段所有特征峰对应的振动模式均源自所有原子参与的集体振动, 主要表现为三氮唑的面外弯曲、 羧酰胺的面外摇摆和呋喃核糖基的弱面内伸缩。 中红外波段, 以晶体结构作为理论计算初始构型的振动光谱比单分子结构计算结果与实验光谱更加吻合, 尤其是400~1 000 cm-1的低频波段, 说明虽然中红外光谱主要来自分子内基团和部分原子的振动, 却也有分子间的相互作用存在, 要想精准的表征特征峰的振动模式, 最好选择晶体结构作为初始计算构型。
结合密度泛函理论和Gaussian16软件, 选择B3LYP杂化泛函和6-311++G(d, p)基组, 对三种抗病毒药物的中红外光谱进行了详细表征, 得到了所有特征峰对应的振动模式。 发现, 2 800 cm-1以下, 振动模式主要源自基团的面内外弯曲和摇摆; 2 800 cm-1以上, 振动模式过渡为C— H, O— H和N— H键的面内伸缩。
我们期望, 该项研究可以为药物生产和存储过程中控制生成目标晶型药物, 抑制优势药物晶型向其他晶型的转化的快速检测提供帮助; 为研究药物的药学特性、 药物间相互作用, 同时, 为解决类似生产工艺等导致药物药效发挥等疑难杂症问题提供借鉴。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|