

{kind=link}
{kind=link}
{kind=link}
{kind=link}
红景天苷分子结构与光谱的密度泛函理论分析
[谢玉玉1, 2, 3  , 侯雪玲
, 侯雪玲1 , 陈志慧2 , 阿吉艾克拜尔·艾萨1, 3, * ]
, 侯雪玲]
|
|


作者简介: 谢玉玉,女, 1984年生,中国科学院新疆理化技术研究所干旱区植物资源与化学重点实验室博士研究生e-mail: xieyy@ms.xjb.ac.cn
随着科学技术不断发展, 中药质量控制和质量评价出现了越来越多的新技术。 在中药标准化过程中, 现代药学研究已经取得了许多令人瞩目的成果, 检测方法和技术取得了很大的进展, 由过去单一指标检测发展到多指标检测, 但由于中草药成分复杂, 作用机制不明等因素制约了中药的发展。 红景天是一种少数民族药, 是中医药发展不可或缺的组成部分。 红景天苷是红景天中的主要有效成分之一, 其提取、 分离和纯化等方法报道甚多, 但其分子键长键角二面角、 前线轨道分布、 表面静电势等分子参数却鲜有报道, 这些都是决定其化学性质和反应机理的重要因素。 采用Gaussian09W化学计算软件的密度泛函理论DFT/B3LYP方法和6-31(d)基组优化红景天苷分子, 得到其稳定结构的键长、 键角和二面角参数。 在此稳定构型的基础上, 计算其表面静电势(ESP)、 最低空轨道(LUMO)、 最高占据轨道(HOMO)、 红外光谱(IR)和核磁共振波谱数据(NMR)。 分别对IR和NMR峰位置进行了归属并与文献报道数据进行了对比, 结果表明: 计算结果的红外吸收频率没有出现虚频说明该优化结果是合理可靠的, 最高占据轨道能量 E=-5.82 eV, 最低空轨道能量 E=-0.000 42 eV, 二者差值为5.81 eV。 通过绘制了轨道的电子云分布图可以看出HOMO轨道主要分布于苯环, 为π电子的成键轨道有一个节面; LUMO轨道也主要分布在苯环上, 为π电子的反键轨道有两个节面。 表面静电势的绘制可以直观地反映出分子中哪部分易发生亲核取代、 哪部分易发生亲电取代反应。 通过绘制第一激发态和基态电子差值, 可以直观地得到电子的迁移方向。 对红景天苷分子计算理论研究可以为进一步探索其化学反应机理、 结构修饰和活性位点的确认等提供重要的参考依据和新思路。
At present, with the continuous development of science and technology, more and more new techniques emerge in quality control quality evaluation of Traditional Chinese Medicines (TCMs). In the standardization process of TCMs, modern pharmaceutical research has made many remarkable achievements. Detection methods and technologies have made great progress from single-index detection to multi-index detection. Rhodiola Rosea is a kind of minority nationality medicine that is an indispensable part of the development of traditional Chinese medicine. Salidroside is one of the main components of Rhodiola Rosea. There are many reports on its extraction, separation and purification. However, far few reports have been reported up to now on its molecular parameters, such as bond length, bond angle, dihedral angle, frontier orbital distribution and surface electrostatic charge Distributions, which are critical factors determining its chemical properties and reaction mechanism. The parameters of bond length, bond angle and dihedral angle of salidroside were obtained by DFT / B3LYP method and 6-31 (d) basis set from Gaussian09W software. As the result of optimization, the surface electrostatic charge (ESP), the lowest occupied orbit (LUMO), the highest occupied orbit (HOMO). Infrared (IR) and nuclear magnetic resonance (NMR) data were calculated, and their peak positions were assigned and compared with the reported data. The results reveal that there is no imaginary frequency in the infrared absorption frequency, which indicates that the optimization result is reasonable and reliable; the highest occupied orbit energy E=-5.82 eV, the lowest orbit energy E=-0.000 42 eV, and the difference is 5.81 eV. By drawing the electron cloud distribution map of the orbit, we can see that the HOMO orbit is the π bonding orbital of electron mainly distributing on the benzene ring with a node; the LUMO orbit is the π antibonding orbital of electron mainly distributing on the benzene ring with two nodes. The drawing of surface electrostatic charge can directly determine which part of the molecule is prone to nucleophilic substitution reaction and which is prone to electrophilic reaction. The electron migration direction can be obtained directly by drawing the electron difference between the first excited state and the ground state. The theoretical study of salidroside molecular calculation in this dissertation can provide important references and new ideas for further exploring the chemical reaction mechanism, structural modification and identification of active sites.
红景天(Rhodiola rosea)又名蔷薇红景天, 为多年生草本植物, 常见于我国的新疆、 西藏和四川, 是一种常见的“药食同源”中药, 有着“高原人参”、 “北极玫瑰”之美称[1]。 经现代药理药效研究证明, 红景天除了具有清肺止咳, 清热凉血和活血止血的作用外, 还具有强壮机体, 抗衰老、 抗疲劳、 抗肿瘤、 抗过敏、 抗抑郁、 耐缺氧、 抗辐射功能和双向调节作用, 具有治疗冠心病和高血压等功效[2, 3, 4, 5]。 红景天是一个具有应用前景的民族药, 值得深入研究与开发。 我国科研人员已经从红景天中提取、 分离并通过FTIR, NMR, MS和UV/Vis等手段鉴定出皂苷类、 多糖类和挥发油等化学成分[6, 7, 8, 9]。 现有红景天的研究主要集中在对有效成分的分离、 纯化、 结构鉴定等方面, 但对其最主要成分红景天苷的几何构型、 电荷分布、 前线轨道和表面静电势鲜有报道。 结构决定性质, 这些都是决定其药理、 药效的重要因素。 随着计算机硬件和软件的开发与利用, 量子化学计算已经在分析化学领域逐渐起到了不可替代的作用, 并逐步应用于药物分析领域。 Gaussian软件是一款常用的量子化学计算软件, 在分子模型构建、 优化分子构型、 计算前线轨道、 表面静电势和模拟红外、 核磁等方面得到了较好的效果, 常用于理论计算与实验测试结果对比和预测活性位点。 本工作采用密度泛函理论, 对红景天苷分子稳定构型、 前线轨道、 表面静电势、 红外与核磁共振等进行了理论模拟和指认, 取得了与文献报道相吻合的结果; 活性位点的指认可以为进一步研究该分子如何诱导肿瘤细胞凋亡、 影响细胞周期、 抑制肿瘤细胞增殖、 增强机体免疫力、 诱导自噬等[10, 11, 12]机理提供了有用的信息和依据。
首先使用GaussView5对红景天苷分子立体结构进行了初始构建如图1(a)所示, 在Gaussian09W程序中, 采用密度泛函理论中的杂化密度泛函B3LYP/基组6-31G(d), 对其初始结构进行优化, 得到红景天苷分子稳定结构, 如表1键长、 表2键角和表3二面角所示。 在优化的基础上, 采用同样的方法和基组计算得出分子的最低空轨道(LUMO)、 最高空轨道(HOMO)、 表面静电势、 红外光谱振动频率和核磁共振的化学位移, 并对计算结果进行了全面指认和详细归属, 取得了与实验数据相吻合的结果。 本项目的全部计算工作通过Gaussian09W程序包在PC机上完成。
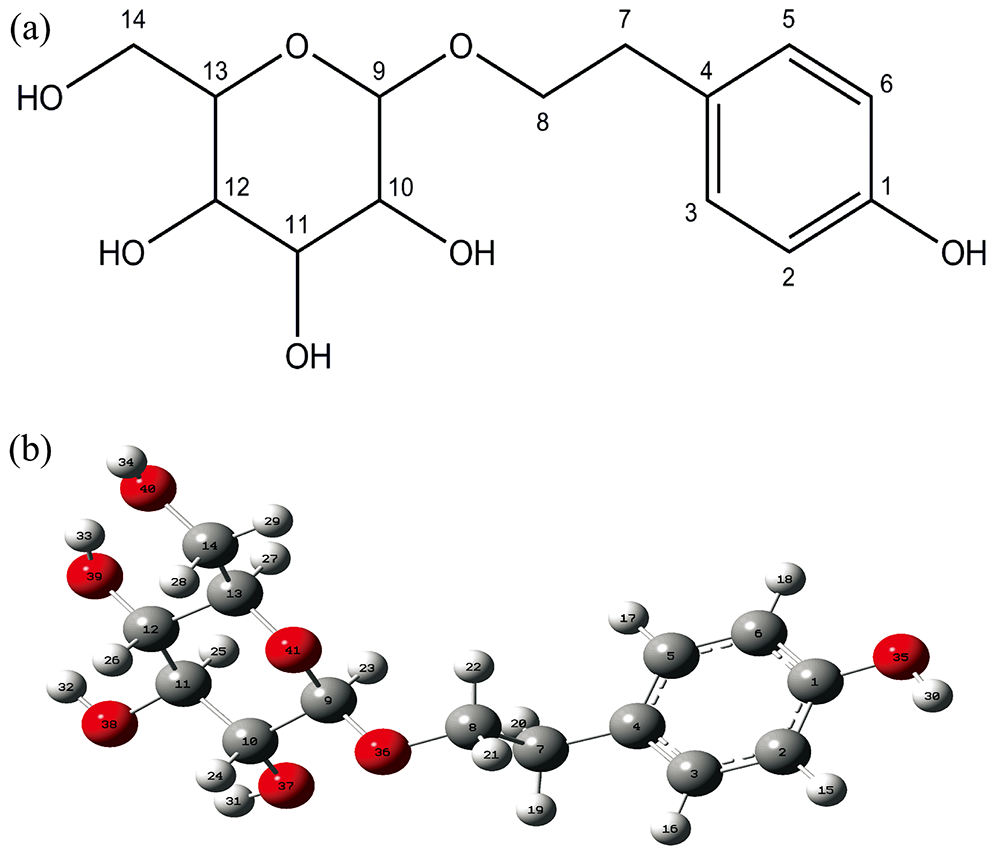 | 图1 优化后红景天苷分子的平面结构图(a)和优化后的立体构型(b)Fig.1 Planar structure (a) and spatial configuration after optimization (b) of Salidroside molecule |
| 表1 Salidroside分子优化后的键长参数 Table 1 Bond lengths of Salidroside after optimization |
| 表2 Salidroside分子优化后的键角参数 Table 2 Bond angles of Salidroside after optimization |
| 表3 Salidroside分子优化后的二面角参数 Table 3 Dihedral angles Salidroside after optimization |
利用密度泛函理论方法优化后的红景天苷分子的立体构型由图1(b)所示。 可以看出该分子的葡萄糖环和苯环并不在一个平面内, 由于苯环上的酚羟基与苯环共平面从而对苯环有所挤压, 3C—4C键长与苯的键长相等为1.4 Å , 4C—5C键长为1.403 Å , 略大于苯环的碳碳键长, 其余均小于苯环的碳碳键长。 糖环上所有的羟基均处在平伏键(e键)上, 符合β -D-吡喃葡萄糖构型。 由于苷元的存在, 葡萄糖吡喃环也有所挤压, 糖环上的C—O—C夹角为114° , 略大于葡萄糖半缩醛上的C—O—C夹角111° 。
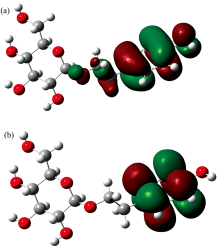
前线分子轨道中的电子是分子中最为敏感的部分, 研究前线分子轨道可以为确定分子活性部位及探讨作用机制等提供信息, 前线分子轨道能量越低, 分子越不容易失去电子, 分子越稳定。 通过Gaussian计算得到Salidroside分子优化后的能量为-29 171.62 eV(-1 072.04 a.u.), 此时分子为稳定状态。 同时计算模拟得出最高占据轨道(HOMO)能量E=-5.82 eV, 最低空轨道 (LUMO)能量E=-0.000 42eV, 能量差值为5.81 eV, 能隙较大, 说明该分子在一般情况下是稳定的。 图2给出了红景天苷分子的HOMO和LUMO轨道的电子云分布图, 由图可知分子的HOMO轨道主要分布在苯环上, 为π 电子的成键轨道, 有一个节面; 而最低空轨道电子云也主要分布在苯环上, 为π 电子的反键轨道, 有两个节面。
 | 图2 (a)红景天分子的HOMO轨道, (b)LUMO轨道Fig.2 HOMO orbital (a) and LUMO orbital (b) of Salidroside molecule |
通过Gaussian09W计算得到Salidroside分子优化后表面静电势, 并投射到分子表面, 如图3(a)所示。 图中红色区域静电势值为负, 表示这一区域更容易给出电子, 或者说相对于其他区域更具有亲核性, 更易发生亲核取代反应; 而蓝色区域静电势值为正, 表示这一区域更容易获得电子, 相对于其他区域更具有亲电性, 易于发生亲电取代反应。 再利用TD-SCF/DFT方法计算第一激发态的电子密度减去基态电子密度, 如图3(b)所示。 蓝色区域表示密度差的正值, 即激发态密度大于基态密度; 红外区域表示密度差的负值, 即激发态密度小于基态密度; 当分子从基态跃迁到第一激发态时, 电子密度从红色区域转移到蓝色区域, 绿色区域的电子密度保持不变。 通过绘制红景天苷分子的分子表面静电势图和第一激发态和基态电子差值, 可以对活性位点进行指认, 从而进一步预测反应机理提供重要的参考。
 | 图3 (a)红景天分子的表面静电势, (b)第一激发态和基态的密度差Fig.3 ESP (a) and Density difference between first excited state and ground state (b) of Salidroside molecule |
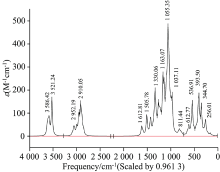
采用Gaussian09W计算出位于中红外区的Salidroside分子红外振动光谱数据, 并根据方法和基组乘以校正因子0.961 3[13], 谱图如图4所示。 分析计算数据, 对红外光谱进行了归属, 在官能团区: 3 640~3 520 cm-1之间为O—H的伸缩振动, 3 100~3 000 cm-1之间为苯环上C—H伸缩振动, 3 000~2 840 cm-1之间为饱和的C—H伸缩振动, 1 612, 1 585, 1 505和1 429 cm-1处为苯环骨架振动, 1 421 cm-1为—CH2—的面内摇摆振动; 在指纹区: 1 256 cm-1处为酚羟基的C—O伸缩振动, 1 137~1 037 cm-1为C—O—C的伸缩振动, 1 032 cm-1为脂肪醇的C—O伸缩振动。 分析以上红外数据发现, 计算结果与文献[14]中报道的红外吸收频率是较为吻合的, 一般情况误差在40个波数内均可认定为吻合。 表4为校正前和校正后部分数据对比, 计算结果并未出现虚频, 故可判断该优化结构合理。
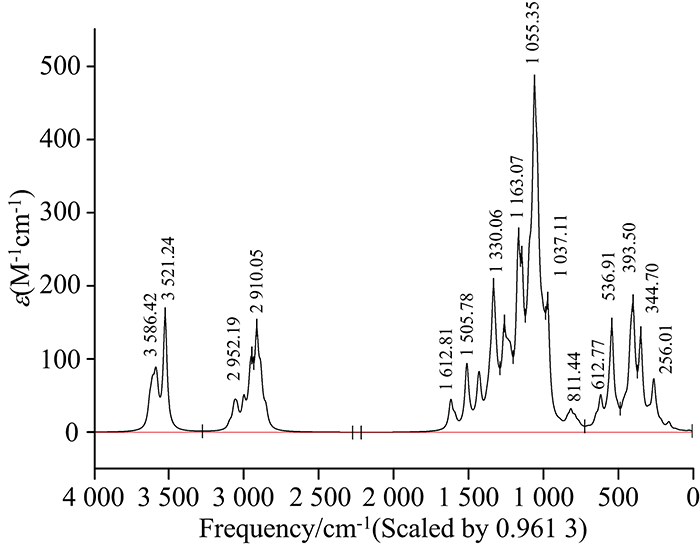 | 图4 优化后的红景天苷分子红外光谱图Fig.4 IR spectrogram of Salidroside molecule after optimization |
| 表4 优化后的红景天苷部分红外振动频率 Table 4 Partial infrared vibration frequency of Salidroside molecule after optimization |
利用Gaussian09W计算出红景天苷分子的13C谱和1H谱, 该分子包含了14个碳原子, 6个为苯环上不饱和碳原子, 其中一个碳原子因为连有酚羟基, 化学位移向低场移至152 ppm, 其他5个碳化学位移在130~110 ppm之间; 1个碳原子为苷元的α -C, 苷化位移向低场移动至64 ppm; 另1个碳原子为β -C受到苷化影响较小, 化学位移在32 ppm; 6个碳为葡萄糖上的次甲基碳原子, 其中一个是糖的端基碳原子, 化学位移向低场移动至91 ppm, 其余5个碳的化学位移在70~60 ppm之间。 同时, 该分子包含了20个氢原子, 4个为苯环上的氢, 化学位移在7.5~6.4 ppm之间; 6个氢为亚甲基的氢, 化学位移在4.2~2.7 ppm之间; 5个氢为葡萄糖次甲基的氢, 其中一个为葡萄糖端基氢, 化学位移在4.21 ppm, 其余4个氢的化学位移在3.3 ppm附近; 剩余5个氢均为羟基的活泼氢。 表5给出了红景天苷分子氢谱和碳谱计算数据和实验数据对比, 文献中均采用CD3OD作为溶剂, 故未给出羟基氢的化学位移。 分析以上数据发现, 氢谱和碳谱计算结果与文献[15]中报道的实验结果基本吻合。
| 表5 红景天苷分子NMR计算数据和实验数据对比 Table 5 Comparison of calculated and experimental data of NMR of Salidroside molecule |
选取民族药红景天中的主要成分红景天苷分子为研究对象, 基于Gaussian09W软件, 采用密度泛函理论DFT/B3LYP方法, 在6-31G(d)基组水平上计算了该分子的稳定几何构型、 红外光谱、 核磁共振谱, 绘制了HOMO和LUMO轨道能量, 并给出了电子云分布图和分子表面静电势图。 结果表明用该方法计算的分子结构是稳定的, 理论计算结果与文献报道结果一致性较好, 计算结果有助于全面了解该分子的结构性质, 为预测其反应机理、 结构修饰、 药理研究等提供理论基础, 对下一步的分子设计具有重要的指导意义。
致谢: 感谢华盛顿大学化学系副主任李晓松老师、 山西大学分子科学研究所吴艳波老师和江苏大学能源研究院李宏平老师在Gaussian软件学习中的帮助和指导。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|