{kind=link}
{kind=link}
{kind=link}
{kind=link}
黄素体系电子转移过程光谱研究
[万浩宇1  , 周子雄
, 周子雄1 , 吴俊彪1 , JörgMatysik2 , 王孝杰1, * ]
, 周子雄]
|
|


作者简介: 万浩宇, 1996年生, 国防科技大学文理学院硕士研究生 e-mail: whychem2020@126.com
黄素类物质在生物体内广泛存在, 是许多电子转移反应的活性中心, 也是电子传递链的重要组成部分。 其受到光照激发后引起的电子转移, 是许多生命过程的基础与起始步骤。 特别地, 一种名为隐花色素的黄素蛋白在光激发后经一系列电子转移形成自旋相关自由基对, 被认为是最有可能的生物磁敏物质, 更使黄素体系电子转移过程的动力学, 特别是自旋动力学过程倍受关注。 对黄素电子转移过程及相关机理进行研究, 有助于厘清多种生命过程的化学机理与影响因素。 为此, 科学界综合运用了多种仪器与测试手段, 其中主要包括紫外-可见光谱, 荧光光谱, 瞬态吸收光谱, 光化学诱导动态核极化(Photo-CIDNP)技术等。 通过多年的研究, 对黄素在生物体内的作用机理与电子转移过程的认识经历了由浅入深, 不断深入的过程。 紫外-可见光谱(UV-Vis)主要用于研究黄素系统中的电子激发, 自旋动力学和电子转移。 结合理论计算, UV-Vis还可以识别电子转移中涉及的基团并进行定量分析。 荧光光谱可以识别电子受激发的物质, 在反应过程中观察黄素和半醌中间体的产生和消耗, 并确定其氧化还原和质子化状态。 瞬态吸收光谱适于观测反应过程中出现的短寿命物种, 其中飞秒泵浦探测技术的引入大大提高了观测的时间分辨率, 并且可以通过光谱特征区分单重态和三重态的自由基对。 光化学诱导动态核极化核磁共振(NMR)可以直接观察电子-核自旋动力学过程。 磁场依赖性photo-CIDNP NMR揭示了控制单重态与三重态互变的因素, 并提出了生物地磁导航可能依赖的化学机制。 腔吸收与单分子光谱的运用, 从技术上提高了实验装置的灵敏度并降低检测限。 主要介绍黄素体系电子转移过程研究所运用的各种光谱手段与取得的成果, 并展望其未来。
Flavins are widely present in organisms and active centers of many electron-transfer reactions. Therefore, they play an important role in biological electron transport chains. Electron transfer caused by light excitation of flavins is the initial step of many living processes. Cryptochromes containing flavin as a cofactor undergo a series of electron-transfer steps to form spin-correlated radical pairs (SCRP) after light excitation. Cryptochromes are considered the most likely candidate for an avian magnetoreceptor, which initiated research on the dynamics of the electron transfer in the flavin system, especially on their spin dynamics. The study of electron transfer and related processes in flavoproteins will allow one to understand biochemical mechanisms and reveal the influencing factors of various living processes. Therefore, numerous research methods, including UV-Vis spectroscopy, fluorescence spectroscopy, transient absorption spectroscopy, electron paramagnetic resonance, photochemical induced dynamic nuclear polarization (photo-CIDNP) and other spectroscopic techniques. We review studies of domestic and foreign scholars on electron transfer of flavin systems, and discuss the recent progress in various major research methods. UV-Vis spectroscopy is mainly used to study electronic excitation, spin-dynamics, and electron transfer in the flavin systems. UV-Vis spectroscopy might identify the groups involved in electron transfer and perform quantitative analysis combined with theoretical predictions. Fluorescence spectroscopy can identify electronically excited species, observe the rise and decay of, for example, flavin and semiquinone intermediates during the reaction course, and identify their redox and protonation states. Transient optical spectroscopy is suitable for capturing short-lived species that appear in the reaction process. In particular, introducing femtosecond pump-probe technology greatly shortened the time-resolution of observation and can distinguish between singlet- and triplet-born radical pair dynamics. Photo-CIDNP nuclear magnetic resonance (NMR) allows -to observe the electron-nuclear spin dynamics directly. Such direct access to the bio-geomagnetic operational mechanism might pave the way for practical applications. Magnetic field-dependent photo-CIDNP NMR reveals the factors controlling the singlet-to-triplet interconversion and suggests a possible chemical mechanism of bio-geomagnetic navigation. The application of cavity absorption and single-molecule spectroscopy technically improves the sensitivity of the experimental device and reduces the detection limit. This article mainly introduces the various spectroscopic techniques to study the electron-transfer process of flavin systems and their research results. Finally, possible future developments in this field are briefly discussed.
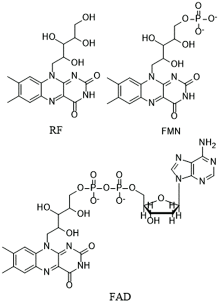
具有7, 8-二甲基-10-烷基异咯嗪结构的黄色化合物通常称为黄素。 其中的成员包括但不限于核黄素(riboflavin, RF), 黄素单核苷酸(flavin mononucleotide, FMN), 黄素腺嘌呤二核苷酸(flavin adenine dinucleotide, FAD)等。
黄素类物质常常以辅基形式出现在蛋白质中, 这些含有黄素辅基的蛋白质被称为黄素蛋白, 它们广泛存在于所有细胞中。 黄素作为一种有机光敏剂, 吸收光子使电子激发, 同时, 它作为电子受体, 可以诱导激发态电荷发生转移。 因此, 黄素是生物体中许多电子转移反应的活性中心, 是电子传递链的重要组成部分。 在对生命过程的研究中, 不可避免地必然会涉及到黄素体系, 所以, 人们很早就对黄素体系的电子转移过程开展了研究。
尤其近年来发现一种称为隐花色素的黄素蛋白[1, 2, 3], 这是一种广泛存在于动植物体内的蓝光受体蛋白, 与DNA光解酶高度同源, 包含一个FAD辅基。 实验表明[4, 5]这种黄素蛋白经光诱导电子转移过程能够形成对地磁场响应的自旋相关自由基对(spin-correlated radical pair, SCRP), 使其成为目前最被认可的生物磁敏物质。 这不仅使生物地磁感应这种神奇的功能可能得到科学的解释, 而且, 其所遵循的自由基对机理(radical-pair mechanism, RPM)是目前唯一能将微弱磁场与化学反应联系起来的理论, 对化学学科的发展也具有重要意义。 因而黄素体系电子转移过程的研究更加受到广泛的关注。
本文主要整理了对黄素类物质及含黄素基团的生物大分子电子转移过程的研究, 对黄素体系电子转移过程的光谱研究手段及其进展作一综述。
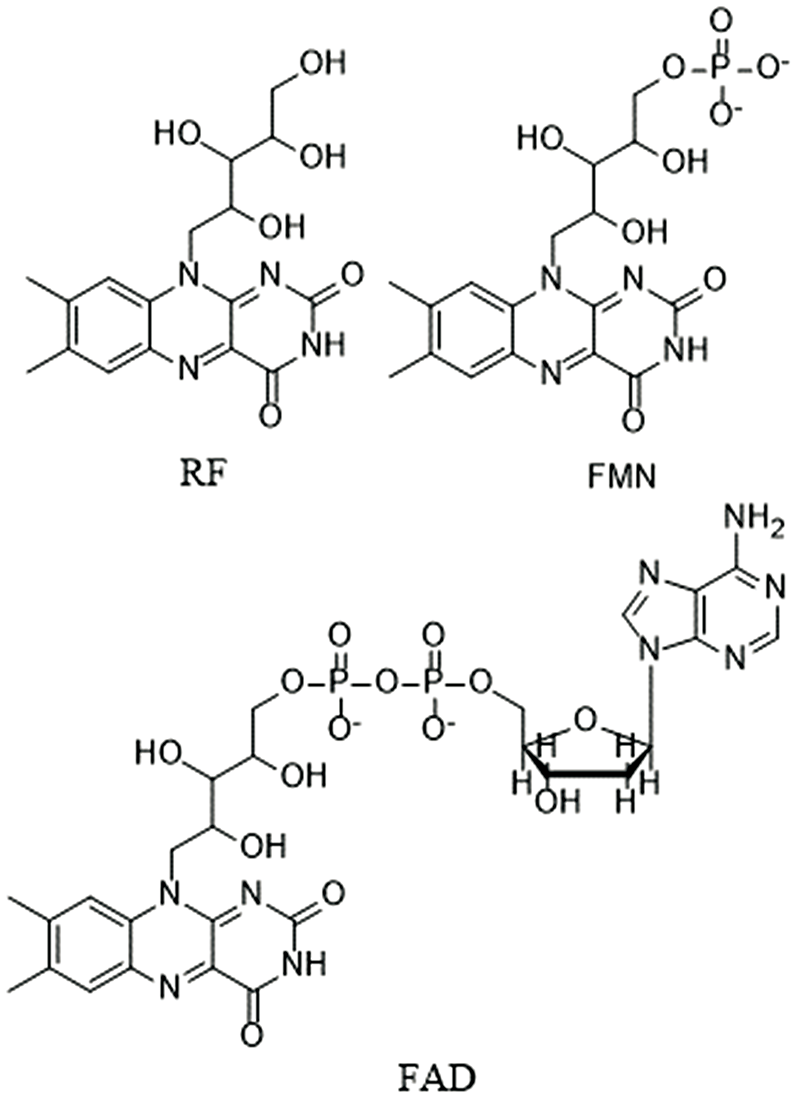 | 图1 核黄素(RF), 黄素单核苷酸(FMN)和黄素腺嘌呤二核苷酸(FAD)的结构式Fig.1 Structural formulas of riboflavin (RF), flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) |
紫外-可见吸收光谱(UV-Vis)是起步较早的一种常用光谱测试手段。 紫外-可见吸收光谱可对分子进行定性和定量分析, 获取其价电子结构及跃迁类型等信息。
黄素中的异咯嗪环(isoalloxazine)是产生UV-Vis吸收峰的主要结构。 早在1965年, Koziol对于核黄素及其光解产物的UV-Vis研究就显示[6, 7, 8, 9], 在蓝光区的440~450 nm处有宽吸收带, 在370 nm附近有一个近紫外吸收带, 分析认为该吸收对应于π → π * 跃迁, 具体数据受溶剂体系的影响而有所差别。 从此开始了对于黄素光激发后分子内电子跃迁过程的研究。
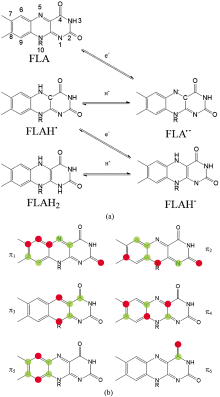
但是, 早期的UV-Vis只能通过吸收光谱判断电子在成键轨道与反键轨道间的跃迁类型, 无法得到关于激发态和电子转移的更多相关信息。 近年来随着仪器技术的发展, 以及计算机技术与模拟计算的引入与运用, UV-Vis又焕发出新的活力。 Mondal等[10]理论计算了黄素生色团的氧化还原状态对UV-Vis的影响, 并用实验进行验证。 发现异咯嗪环上的π 3分子轨道主要影响了黄素类物质中异咯嗪环的光物理性质, 其上的N5=C4a=C10a=N1键在还原过程中转变为HN5=C4a=C10a=N1H的形式(如图2), 从而使得黄素分子可以作为电子受体接受电子。
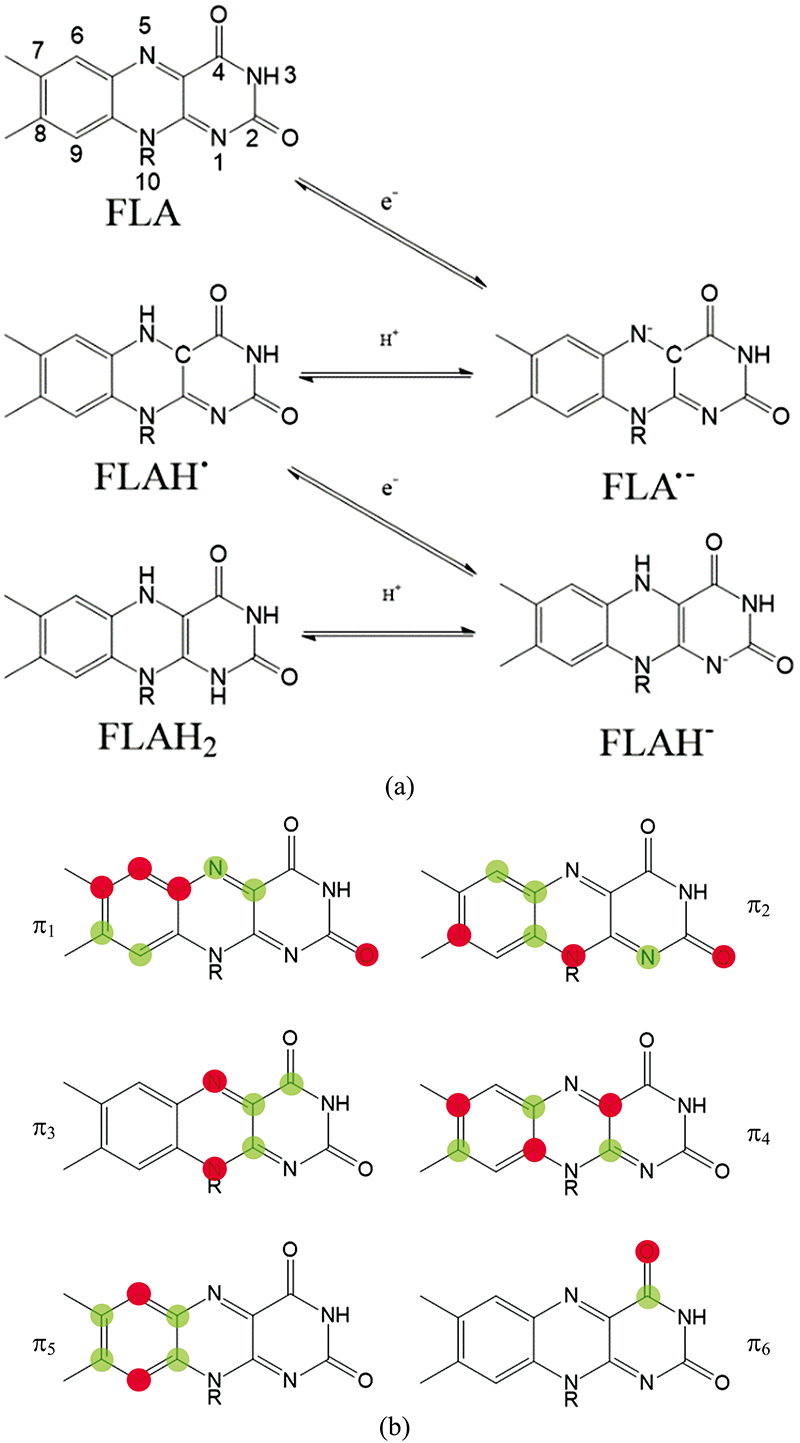 | 图2 黄素几种氧化还原态(a)与π 键分子轨道(b)示意图Fig.2 Schematic diagrams of several redox and protonation states of flavin (a) and π bond molecular orbitals (b) |
Karno[11]等通过先模拟计算, 后实验验证的方式, 研究了拟南芥隐花色素蛋白中FAD及其另外四种衍生化合物的UV-Vis, 在FAD· -和 FADH· 的吸收光谱中找到了酪氨酸和色氨酸自由基的特征峰, 并推算出物种比例为1∶ 1, 支持了酪氨酸残基也参与了隐花色素蛋白电子传递链的猜想。 Kar[12]等用量子化学方法计算了光黄素的电子结构和光谱性质。 用各种基于波函数的方法和密度泛函计算了10个单重态和三重态的激发能, 并分析了电子密度的差异。 在耦合团簇方法CC2的基础上, 建立了单重态和三重态激发态的相对级数。 发现至少需要7个单重态激发态才能指定UV-Vis光谱中的所有峰。
UV-Vis从早期的通过简单识别特异性吸收带判断电子跃迁类型进行定性分析, 发展到与理论预测相结合, 精准识别参与电子传递的基团并进行定量分析。 可见, 紫外-可见吸收光谱作为应用最广泛的光谱研究手段, 历久弥新, 仍然在黄素电子转移机理的研究中有着重要的地位。
荧光是辐射跃迁的一种, 是物质从电子激发态跃迁到低能状态时所释放的辐射。 研究黄素及其衍生物的荧光光谱, 有助于研究受激后的光循环过程及其影响因素。
早期的荧光光谱研究内容比较简单, 1974年Ghisla等[13]测定了一系列还原黄素蛋白的荧光和吸收性质, 并与经取代修饰过的光黄素进行了比较。 发现还原黄素生色团对溶剂的极性、 pH值以及粘度的变化和R基的取代非常敏感, 对之前观察到的吸收光谱和荧光光谱变化进行了解释。 Tyagi[14]等测试了光黄素在水溶液中吸收和发射行为的pH依赖性, 发现不同pH值下光黄素以不同的离子状态存在, 基态与激发态势能曲线交叉方式有所不同。 荧光发射行为和吸收行为的pH依赖性一致。
Penzkofer[15]从黄素中的异咯嗪环结构出发, 利用荧光光谱研究了咯嗪, 异咯嗪, 向光素(7, 8-二甲基咯嗪)及其同分异构体7, 8-二甲基异咯嗪, 发现咯嗪的最高占据n轨道(n-HOMO)的能量远低于最高占据π 轨道(π -HOMO)的能量, 但其余三种物质的n-HOMO能量仅略低于π -HOMO, 从而在光致激发进行π → π * 跃迁后发生了n→ π * 电子转移, 延长了荧光寿命。
由于荧光光谱所能观察到的时间尺度远小于UV-Vis, 故能更好地分辨电子传递过程中结构变化引起的光谱变化。 如Shirdel等[16]研究了黑腹果蝇昼夜蓝光感受器隐花色素(dCry)在暗适应(受体)状态、 光适应(信号)状态和长时间紫光照射下的吸收和荧光行为。 dCry中的FAD辅因子以氧化形式(FADox)存在。 U形FADox的光激发引起从腺嘌呤部分到异咯嗪部分的快速分子内电子转移, 紧随其后的是从相邻色氨酸残基到带正电腺嘌呤部分的分子间电子转移。 恢复了中性的腺嘌呤部分充当了异咯嗪阴离子和相邻色氨酸阳离子之间的电荷分离剂。 电荷分离, 蛋白质构象变化和蛋白质极化引起的电荷屏蔽使FAD· -在极短的时间范围内保持稳定, 缓慢地重新氧化成FADox。 FAD· -也能再受光激发引起氧化电子转移, 从而在皮秒至纳秒的时间尺度上将FAD· -转换回FADox, 并将Trp+转换为Trp。 初步描绘了黄素体系光化学循环过程。 Insinń ska-Rak[17]报道了四种核黄素衍生物(5DRfl, IRfl, 3MeTARF和3BLfl)在甲醇中的光诱导降解过程。 吸收光谱与发射光谱研究发现光解过程具有相当大的量子产率。 其中5DRfl和IRfl在厌氧条件下进行的光解比在氧气存在下进行的光解要有效得多, 说明三重激发态参与了光化学过程。 唯一的例外是3BLfl, 其中没有检测到氧对光解稳定性的影响, 因为氧的存在不影响3BLfl的光解量子产率。 对3-苄基-核黄素和3BLfl获得的结果的比较证实光降解主要发生在核糖基链。 无论存在或不存在氧, 该反应仍以相似的量子产率发生。 因此, 该反应很可能不涉及三重态。 与其他黄素相比, 3MeTARF则明显更耐光, 其衍生物的光稳定性高于核黄素本身。
但是, 仅仅对于黄素及衍生物的荧光光谱进行研究, 而不考虑其在生物中实际的作用机制, 显然欠缺一定的说服力。 荧光光谱同样可以针对生物对象直接开展研究。 Hoang等[18]使用杆状病毒衍生的表达系统, 在活的Sf21昆虫细胞中高水平表达了人和果蝇的隐花色素蛋白。 使用蓝光照射完整的细胞, 通过荧光和电子顺磁共振波谱监测产生的隐花色素光反应过程。 发现人类和果蝇细胞内隐花色素蛋白中结合的黄素, 在光诱导下其氧化还原状态发生改变。 黄素氧化态的还原与半醌中间体信号的出现同之前从植物中提取的隐花色素蛋白在体外观察到的变化相同。 表明隐花色素在生物体内外所经历的光反应过程是一样的。
荧光光谱能够识别物种的基态与激发态, 并且时间尺度更短, 非常适合研究反应过程中出现的激发态中间体。 通过荧光光谱, 黄素体系光反应过程中的多种中间体被识别出来, 光反应过程被补充完全, 有力地促进了黄素电子转移过程的探索。
瞬态吸收光谱(TAS)是一种常见的超快激光泵浦-探测技术, 是研究物质激发态能级结构及激发态能量弛豫过程的有力工具。 将瞬态吸收光谱应用于黄素光循环过程研究, 能够捕捉到中间产生的自由基对等物种。 相比于稳态光谱, 瞬态光谱的分辨能力能够达到飞秒甚至更低的时间尺度。
2003年Giovani[19]等率先利用瞬态吸收光谱证明拟南芥隐花色素的初级光反应涉及从色氨酸和酪氨酸残基到激发的FAD辅基的蛋白内电子转移。 2008年Lukacs[20]利用瞬态吸收光谱研究了大肠杆菌DNA光解酶的光致电子转移, 发现电子在三个色氨酸残基间的转移速度较快, 在约30 ps的时间内沿残基链跳跃, 使FAD辅基形成还原态的FADH-, 但是FADH--Trp· +的稳定依赖于色氨酸的去质子化, 时间尺度在200 ns。 Li等[21]用飞秒瞬态吸收光谱研究了FAD和FMN的激发态动力学随pH的变化。 他们表征了黄素在三种质子化状态下的激发态吸收光谱, 发现阴离子和中性状态具有相似的激发态吸收光谱, 但阴离子单重激发态系间窜越率远高于中性激发态。 质子化激发态的瞬态吸收光谱明显不同于其他形式, 这表明质子化后发色团中存在高度的电子再分布。 对质子化激发态的进一步研究发现基态和激发态分子呈现不同的质子化位置。 在低pH和高pH条件下, FAD均为“ 开放” 构象, 其激发态动力学与FMN相同。 这种构象变化是由于腺嘌呤环在低pH时质子化和异咯嗪环在高pH时去质子化而引起的。 在中性pH范围内, FAD采用“ 闭合” 构象, 由于两个芳香族部分之间的堆积作用, FAD表现出快速的激发态失活。
同时, 由于仪器技术的进步, UV-Vis的测定也开始向时间分辨领域进行拓展, 观察到一些寿命较短的物种对吸收光谱的影响。 2009年Langenbacher[22]引入时间分辨UV-Vis技术, 以藻类隐花色素作为研究对象, 确认从色氨酸到黄素的电子转移在100 ns内完成, 形成了黄素阴离子自由基, 而发生质子转移的物种衰变时间常数则为1.7 μs。 将电子转移和质子转移的时间尺度严格区分开。 2010年的进一步研究表明, 300 ps~10 ns的时间尺度内几乎没有吸收变化, 说明此范围内没有电子的转移与重新结合[23]。 同年Brazard[24]利用宽带UV-Vis飞秒吸收光谱对隐花色素进行了研究, 发现色氨酸链的电子转移速度比在DNA光解酶中快, 已经达到亚皮秒尺度。 原因可能是在隐花色素中, 黄素基团的存在形式是氧化态的FADox, 其最低激发态的电荷分布有利于近端色氨酸残基的电子转移。 最近, Ernst[25]监测了一种黄素蛋白— — PqsL单加氧酶经历光诱导还原过程的时间分辨UV-Vis, 阐明了NAD(P)H和FAD之间的电荷转移涉及一个单电子转移的两步反应, 导致了一个瞬时的中性半醌中间体的生成, 证实了蛋白中黄素的完全还原是通过两个连续的电子转移步骤, 而不是通过离子的一步转移完成的。 Antill[26]等设计仿生囊泡系统研究了黄素与色氨酸在脂质双层膜上的电子转移反应, 在微秒级别的时间尺度上, 对于三种不同的囊泡系统, 测定了UV-Vis, 推算了动力学过程。 证明嵌入囊泡双层中的核黄素四丁酸酯(RFTB)可以与色氨酸分子在囊泡内部或整体溶液中进行电子转移。 包裹在囊泡内部的FMN可以经过小泡双层进行电子转移, 从而产生磁性敏感的自由基对, 扩散重遇产生的自由基对之间还可以通过借助简并电子的交换能发生电子反向转移。
Zirak[27]利用瞬态吸收光谱比较了玫瑰黄素和核黄素在水和有机溶剂中的光动力学, 发现与核黄素不同, 玫瑰黄素的激发态分子内电荷转移是通过二甲氨基的给电子效应和羰基的吸电子效应来实现的。 Chang等[28, 29]则从结构出发, 首先研究了FMN和FAD在溶液和惰性蛋白质环境中五种氧化还原状态下的激发态动力学, 观察到黄素寿命和电子转移动力学受异咯嗪环的平面性与柔性控制, 之后在皮秒尺度观测了黄素蛋白三个氧化还原状态中的超快溶剂化动力学, 阐明了水合网络和局部蛋白质构象之间的内在动力学关系, 最终将影响黄素蛋白的电子转移过程。
随着仪器技术的发展, 飞秒泵浦-探测技术也开始用于化学研究, 尤其在超快动力学领域大放异彩。 2012年Immeln等[30]运用了飞秒泵浦-探测技术, 研究隐花色素受蓝光激活后的蛋白分子内电子转移, 将电子从色氨酸向黄素基团转移的初级过程确定在0.4 ps的时间尺度。 2014年Mü ller等[31]研究了ATP在植物黄素电子转移中的作用, 发现初级电子转移形成FAD· --Trp1H· +自由基对后, 后续电子转移的路径分配受pH值和ATP的影响。 ATP会降低质子与FAD辅基的结合能力, 促进色氨酸电子传递链与质子转移的竞争。
超快光学手段也能够分辨出一些之前观察不到的动力学过程。 单重态与三重态吸收光谱发生的红移程度不同, 光谱的时间分辨率越高, 越容易将二者区分开。 例如尽管早期证据和理论支持隐花色素中的自由基对起源于三重态[32], 但之后观测到的隐花色素中的磁敏自由基对都起源于单重态[33], 这一问题长期困扰着科学界, 一种可能是三重态的特征吸收被单重态掩盖难以分辨。 2019年Bialas[34, 35]等在四螺旋蛋白质骨架上固定黄素和色氨酸残基, 运用飞秒泵浦-探测方法, 测量了其黄素-色氨酸自由基对光致电子转移的动力学过程。 首次确认了黄素-色氨酸自由基对的形成既可以始于单重态, 也可以始于三重态。
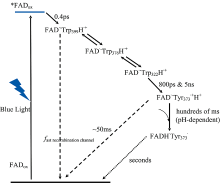
Martin[36]等用飞秒偏振瞬态吸收光谱对非洲爪蟾(6-4)光解酶中带有氧化态FAD的光活化反应进行了详细的分析。 非洲爪蟾(6-4)光解酶的特点是其电子传递链上有四个而非三个色氨酸残基。 实验表明被激发的黄素约0.5 ps内被附近的色氨酸残基还原, 产生FAD· -和TrpH· +自由基。 在皮秒时间尺度下的后续动力学步骤中, 正电荷沿着色氨酸四联体进行迁移, 与电荷重组竞争。 并且观察到第四个色氨酸的氧化速度与第三个色氨酸的氧化速度一样快(约40 ps), 这意味着这两个残基之间的电子转移速度非常快。 由此他们提出, 在大多数动力学过程中, 正电荷实际上在各种色氨酸残基上离域化, 而电荷重组是通过近端色氨酸残基发生的。 Lacombat[37]则报道了传递链上第四个氨基酸为酪氨酸的莱茵衣藻类动物隐花色素(CraCRY), 其FAD辅基还原与色氨酸三联体传递电子的时间尺度与其他隐花色素类似, 但酪氨酸残基的氧化和去质子化则在约800 ps内发生, 使得FAD· -更易质子化为长寿命的FADH· -物种, 延长了自由基对的寿命(图3)。
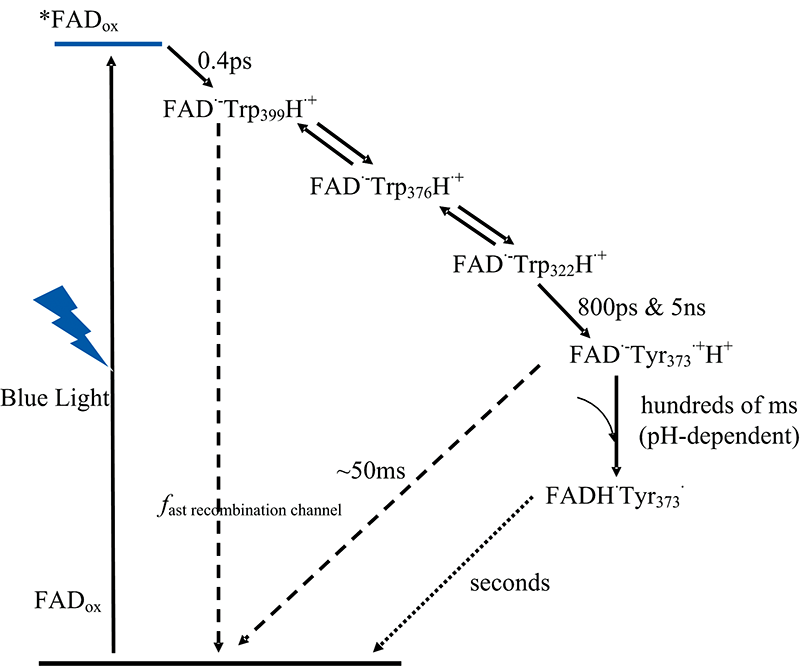 | 图3 莱茵衣藻(CraCRY)隐花色素电子转移过程[37]Fig.3 The electron transfer process of the animal-like cryptochrome of Chlamydomonas reinhardtii (CraCRY)[37] |
瞬态吸收光谱还可用于化学磁效应的相关研究, Kerpal等[38]使用瞬态吸收光谱证明CPF分子三联体系统在与鸟类磁接收相关的场区中的磁场响应具有明显的方位依赖性。 这些结果强调了基于自由基对的化学磁罗盘的可行性, 为可开发的化学磁罗盘系统的设计和操作提供了进一步的指导。 随后Lewis等[39]证明, 施加弱磁场的条件下, “ 低场效应” 会导致自由基对经历S和T0态之间依赖于时间的布居转移的新路径, 而不是先前所认为的S和T± 态之间的布居转移。 解决了当与“ 高场效应” 相结合时, 如何解释在某些磁场强度下观察到的在CPF自由基和瞬态吸收实验中观察到的不寻常的三相行为的问题。
作为目前最为先进的光学研究手段, 瞬态吸收光谱能够在皮秒乃至飞秒的时间尺度精确地识别目标物种, 分辨动力学过程的时间顺序。 其应用取得了许多新的成果, 尤其对自旋相关自由基对的研究, 极大地推进了黄素电子转移过程研究进程。 但也因其出现较晚, 在实验平台的搭建和实验的设计上还没有较为成熟的通用方案, 仍有较大的改进空间。
光化学诱导动态核极化(photo-chemically induced dynamic nuclear polarization, photo-CIDNP)是一种在光照条件下由于产生非玻尔兹曼核自旋极化而使核磁共振(NMR)波谱信号强度发生明显变化的效应。 核磁共振(NMR)谱可以观察到这种非玻尔兹曼核自旋极化增强的吸收(正)或发射(负)信号。 发生变化的NMR信号为人们深入了解自由基反应中间体提供了便利条件。 因此, photo-CIDNP技术成为研究光诱导电子转移反应机理和反应中间体的十分有效的手段。
液态photo-CIDNP效应最早在1967年被发现[40, 41]。 其发生机理是自由基对机理(RPM), photo-CIDNP信号强度与局部电子自旋密度(各向同性超精细耦合常数aiso)相关, 并且信号的符号取决于反应参数, 例如超精细耦合常数HFC, 朗德因子g等。 液态photo-CIDNP的磁场依赖性实验允许对所有这些参数进行建模, 从而确定自旋相关自由基对(SCRP)的自旋动力学过程。 2005年Richter等[42]以紫花苜蓿向光素LOV2 450A突变体为研究对象, 首次报道了用液态NMR探测的完整黄素辅因子-蛋白质体系中的photo-CIDNP结果。 紫花苜蓿向光素中含有FMN辅基, 在蓝光照射下, 在属于FMN的异咯嗪环部分的某些碳的共振中出现了异常强烈的核自旋极化。 相反, 属于FMN核糖基侧链碳的NMR信号强度不受光的影响。 证明光照激发后参与生成自由基对中间体的是FMN中的黄素部分。 2014年Kothe等[43]对同样的突变体进行研究, 确认其中photo-CIDNP效应的出现来自于黄素受激后的三重态及形成的三重态自由基对。
Feldmeier等[44]开发了一种用于NMR光谱研究的LED照明装置, 能够将样品内部的光强提高约800倍。 这种LED装置可以在脉冲和连续波模式下使用, 并且便于时间分辨的photo-CIDNP以及整个反应路径的检测, 可以用来研究光催化中下游中间体和溶剂效应的相关性。 为photo-CIDNP的进一步发展提供了技术支持。 Pompe等[45]详细分析了水环境中FMN和三个具有不同甲基化模式的类似物的时间分辨photo-CIDNP谱, 根据在不同pH值下记录的photo-CIDNP谱, 推导了所研究的黄素类似物与L-色氨酸的光反应的机理细节。 这为研究某些蓝光感受器的电子转移机理以及探究用黄素类似物取代天然黄素的方法开辟了新的途径。
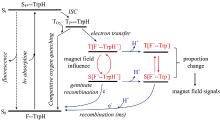
王孝杰课题组[46, 47, 48]合成了黄素-色氨酸二联体分子F10T, 其中黄素(F)残基通过脂肪链与色氨酸(Trp)残基相连。 黄素和色氨酸之间中心到中心的距离(约16.5 Å )与天然隐花色素中FAD和色氨酸之间的距离(约18 Å )接近。 液态1H Photo-CIDNP NMR研究发现F10T光化学过程同FMN/Trp溶液体系一致, 表明F10T保持了分子各部分性质独立, 具有超分子特性。 其photo-CIDNP信号不具有浓度依赖性, 表明F10T发生的是分子内电子转移。 在蓝光照射下, F10T受激发后经电子转移形成SCRP通过磁场依赖性photo-CIDNP研究发现, F10T形成的SCRP单重态与三重态之间的转化可以被极微弱的磁场影响(如图4), 从而开创了通过模型化合物开展生物磁感应研究的新途径, 为化学磁罗盘的开发提供了新的思路。
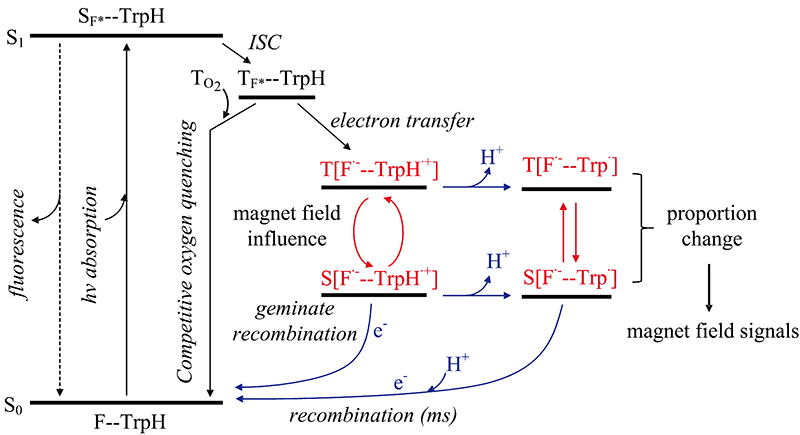 | 图4 F10T分子受外加磁场影响的光循环过程[48]Fig.4 The photochemical cycle process of F10T molecules affected by an external magnetic field[48] |
由于液体核磁共振研究大分子量、 高粘度样品时存在局限性, 学界开始研究在固体魔角旋转(magic-angle spinning, MAS)核磁共振中是否存在photo-CIDNP效应。 固态和液态photo-CIDNP之间的一个显著差异是, 液态photo-CIDNP的发生机理是自由基对机理(RPM), 而固态photo-CIDNP除了RPM外, 还有与RPM一同起作用的另外三种机制: 三旋混合(TSM), 衰变差异(DD)和弛豫差异(DR)。 已有研究者认为, TSM是禽类生物磁罗盘自旋变化的重要模式。 与RPM相似, TSM极化是通过三重态和单重态之间的相干振荡来放大的。
固态photo-CIDNP效应首次在冷冻的细菌光合作用反应中心(reaction centers, RCs)中被观察到[49, 50], 之后在所有的天然光合作用RCs中都观察到了固态photo-CIDNP效应, 但在光合作用RCs以外的系统中很长一段时间内都没有发现这种效应。 直到2010年Jö rg等[51, 52]于向光素(LOV1-C57S)的突变体中首次观察到非光合作用系统的固态photo-CIDNP效应。 为研究黄素电子转移提供了新的研究手段。 并且, 固态photo-CIDNP信号在外加磁场的作用下出现了明显的差异, 为探究鸟类地磁导航的生物学机制提供了新思路和方法。 研究表明, 隐花色素的固定化会导致HFC的张量性质在磁场上产生各向异性相互作用, 从而形成磁罗盘的基础架构。
近年来, photo-CIDNP向利用同位素标记特定位点研究光反应结构域进行拓展, Ding等[53]用同位素标记了色氨酸电子供体。 研究了莱茵衣藻的半胱氨酸缺失型向光素LOV1-C57S中的固态photo-CIDNP效应及其机理, 不仅观察到了13C photo-CIDNP信号, 而且首次在缺乏半胱氨酸的结构域中观察到了15N photo-CIDNP信号。 随后测量了磁场依赖性, 理论能级交叉分析表明, 在强磁场下, 各向异性机制起主导作用, 这意味着在溶液中出现了与固态photo-CIDNP相同的机制。
photo-CIDNP技术将黄素体系电子转移过程的研究向自旋动力学领域进一步拓展, 能够测定自旋相关自由基对在环境影响下产生各向异性相互作用的变化过程, 对电子转移过程中有自旋相关自由基对产生的反应体系尤为适合。 并且photo-CIDNP能够反映出低至地磁场水平的微弱磁场变化, 将为黄素体系参与生物地磁导航机制的研究及化学磁罗盘的开发提供有力的帮助。
通常情况下, 光谱学研究的是体系内全部分子的统计数据, 实际上得到的是整个系统的平均响应或某个物理量的统计平均值, 这一平均效应掩盖了许多特殊的信息。 如果尽可能降低被测样品浓度, 乃至针对单个分子展开研究, 就能够更好地观察反应过程, 从而得到与分子微环境相关的信息等。 但是显然, 样品浓度的降低会带来信号强度减弱, 噪声过大等问题, 需要提高实验装置的灵敏度并降低检测限。 为此, 腔吸收光谱与单分子光谱技术走进了化学家的视野。
腔吸收光谱主要包括腔衰荡吸收光谱(CRDS)与腔增强吸收光谱(CEAS)。 二者都需要将样品置于激光谐振腔内, 不同之处在于腔衰荡吸收光谱通过测量谐振腔内激光的衰减速率来反演分子浓度, 对机械斩波的精确度要求较高; 腔增强吸收光谱是基于腔衰荡吸收光谱技术发展起来的一种新的高灵敏度光谱技术, 通过扫描激光频率, 使其与谐振腔某一腔模频率共振而激发腔内光场。 Zollitsch等[54]设计了腔衰荡的瞬态泵浦-探测实验装置, 使用微型泵控制进样量, 以人工合成的黄素蛋白为对象研究了隐花色素中黄素与第一个色氨酸残基之间的初级电子转移过程, 发现色氨酸残基与黄素残基的距离与磁敏性之间存在强相关。 由于人工合成蛋白中二者距离较天然蛋白要近, 电荷分离不能有效地与系间窜跃竞争, 其结果是其中的自由基对以三重态而不是单重态产生, 并表现出与天然隐花色素相反的磁场效应。
单分子技术不仅可以用于吸收光谱和荧光检测, 也可以进行EPR和NMR实验, 但是对于具有超分子性质的自由基对是否适用还没有确切的结论。 Ikeya等[55]尝试将单分子光谱的范围扩展到单个自由基对, 对利用FAD分子进行自旋化学实验的可行性进行了详细的研究。 根据理论预测提出了单个自由基对的单分子光谱的一致性理论, 表明聚集现象受自由基对中单重态-三重态相互转化的影响, 而相互转化又受局域磁场和外磁场的影响。 结论是, 通过荧光检测观测单个FAD分子的磁场效应是可行的, 但在实验上对综合荧光强度和荧光事件统计的测量具有挑战性。
不难看出, 具有高灵敏度的腔吸收光谱与单分子光谱在研究黄素体系电子转移中有着极为广阔的应用前景, 但是由于设备要求高, 起步较晚, 还有一些理论与实验中的难题尚未解决, 有待进一步探索。
黄素类物质常常在生物体内充当蓝光受体, 研究其受光照激发及后续的电子转移过程显然对于人们深刻认识多种生命过程的化学机理有着重要的意义。 光谱手段从早期的单纯根据吸收峰判断电子跃迁类型, 到后来的通过瞬态光谱分辨物种和反应历程, 再到与波谱手段联用检测化学环境最细微的变化, 与相关技术和理论的进步是分不开的。
总体来看, 黄素体系电子转移过程研究主要集中在两个方面: (1) 光化学反应过程基本规律研究。 早期研究集中于此, 也涉及到了一些动力学过程。 主要利用的是光谱手段, 涉及光化学过程。 从整体上看, 呈现时间尺度由长到短, 灵敏度由低到高, 分辨率由粗糙到精确的特点。 尤其是超快光学手段的引入, 使得时间分辨率有了大幅的提高。 当然, 光谱信息纷繁复杂, 如何解读光谱信号, 从中提取有用的信息也是一个需要认真思考的问题。 尤其值得注意的是, 新兴的光谱手段还没有一个比较通用的实验方案, 基本是根据实际情况来设计实验装置与方案, 在可重复性和规范性上还有所欠缺。 对此, 学习物理学中一些成熟的技术会是比较好的选择。 同时, 由于黄素体系普遍具有易光解的特点, 还需要在光斑能量密度等方面做出适合光化学研究的调整。 (2) 生物地磁感应机理研究。 隐花色素作为生物磁敏物质的观点被提出后, 它的电子转移过程不仅涉及生物地磁导航的机理, 还涉及化学磁场效应的实现。 故而后期研究更关注于此, 拓展到自旋动力学领域, 尤其是对于其中形成的自旋相关自由基对的研究。 这一时期在原有技术的基础上结合运用了多种波谱手段, 取得了较大的进展。 通过这些新手段的引入, 有望能更加清楚地阐释黄素体系电子转移过程在生物地磁感应机制中扮演的角色, 推进化学磁罗盘的开发。
本文主要介绍了黄素体系电子转移机理研究过程中所运用的各种光谱/波谱手段与取得的研究成果, 对其未来发展方向进行展望。 我们相信, 随着技术的发展与理论的完善, 加之科研工作者的不懈努力, 在不远的将来黄素电子转移机理的面纱将被完全揭开。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|