




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
深紫外分光光度检测系统的稳定性及灵敏度研究
[张雪菲1  , 段宁
, 段宁1, 2, * , 降林华1, 2, * , 程雯2 , 于兆胜3 , 李维栋2 , 朱广彬4 , 徐艳丽2 ]
, 段宁, 降林华, 程雯|
|


作者简介: 张雪菲, 1995年生, 安徽理工大学材料科学与工程学院博士研究生 e-mail: 470066547@qq.com
现有的国标光度法无法直接测定流程工业中连续反应单元生产过程的污染物, 主要原因是氧气在深紫外区对紫外光的吸收干扰了紫外分光光度计对目标物质的检测, 导致检测结果存在一定程度偏差。 因此, 解决这一问题的关键核心是稳定获取深紫外区不同特征波长物质的高灵敏光度信息。 在紫外分光光度计基础上加装氮气输配系统, 同时设计了自动进样流通池及进样托盘以实现检测间隙自动进样功能, 减少检测间隙氮气消耗。 为提高仪器稳定性, 分别精准控制通入仪器内部光学系统区、 样品室和数据接收区三个腔体的氮气流量, 数值分别为6, 2和3 L·min-1, 使仪器基线平直度平均值由0.108降低至0.010, 较空气条件削减了90.7%。 通过对比空气与氮气两种气氛下直接测定SO42-的吸光度、 灵敏度、 灵敏度变化量和线性范围的差异, 发现氮气气氛下检测结果的吸光度和灵敏度在光程 b=1~100 mm范围内均有提升, 灵敏度变化量随 b=1 mm时的10.42%增大至 b=100 mm时30.65%, 线性范围却随光程的增加由0.09 g·L-1缩短至0.03 g·L-1。 说明氮气输配系统能够成功抑制检测过程中紫外光强度的衰减。 与检测SO42-的常用方法之一的离子色谱法相比, 该方法具有检测便捷、 检测结果稳定可靠并且经济效益良好的优势, 可为工业实际应用奠定基础。
, DUAN Ning, JIANG Lin-hua, CHENG WenThe existing national standard photometric method can not directly determine the pollutants in the production process of continuous reaction units in the process industry. The main reason is that the absorption of ultraviolet light by oxygen in the deep ultraviolet region interferes with the detection of target substances by ultraviolet spectrophotometer, resulting in a certain degree of deviation in the detection results. Therefore, the key to solving this problem is to stably obtain the highly sensitive photometric information of substances with different characteristic wavelengths in the deep ultraviolet region. In this study, a nitrogen transmission and distribution system was installed based on a UV spectrophotometer. At the same time, an automatic injection flow cell and tray are designed to realize automatic sample injection between detection gaps. The nitrogen flows into the optical system area, sample room and data receiving area of the instrument is accurately controlled at 6, 2 and 3 L·min-1 respectively, so that the average value of the baseline flatness of the instrument is reduced from 0.108 to 0.010, which is 90.7% less than the air conditioner. Comparing the differences in the absorbance, sensitivity, sensitivity change and linear range of SO42- between air and nitrogen atmospheres, reveals that the absorbance and sensitivity of detection results in nitrogen atmosphere are improved in the range optical pathlengths b=1~100 mm. The sensitivity change increases from 10.42% to 30.65% when b=1 mm to b=100 mm, but the linear range decreases from 0.09 to 0.03 g·L-1 with increasing of optical path lengths. It shows that the nitrogen transmission and distribution system successfully inhibits the attenuation of UV intensity in the detection process. Compared with ion chromatography, one of the common methods for detecting SO42-, this method has the advantages of convenient detection, stable and reliable detection results and good economic benefits, laying a foundation for industrial application.
对污染物进行实时监测是实现清洁生产的基本前提。 工业过程液相体系中的反应速度普遍较快, 而现有的国标分光光度法在检测前需要对样品进行稀释定容和络合显色等预处理[1, 2, 3, 4, 5, 6], 因此检测结果相对工业液相体系实时状态存在严重滞后, 难以实现对工业过程实时反馈调控。 导致这一问题的主要原因是氧气在紫外波段内具有强吸收特性[7, 8], 易吸收波长< 240 nm的紫外光。 当氧分子吸收紫外光后, 氧分子间的化学键被打断, 光致离解成一个基态氧原子O(3P)和一个电激发态氧原子O(1D), 基态氧原子O(3P)又是形成臭氧分子的重要反应物。 臭氧分子也会因吸收200~350 nm波段的紫外光使臭氧分子结构被破坏[9]。 因此, 氧气与臭氧之间的可逆转化不仅会消耗紫外光能量[8], 而且氧气与臭氧均吸收一定量的紫外光, 影响检测结果的稳定性与准确性, 导致难以准确直接测定深紫外区物质。
破解现有光度方法不能直接测定流程工业连续反映单元生产过程中污染物的难题, 其关键核心是稳定获取深紫外区不同特征波长物质的高灵敏光度信息。 为隔绝氧气干扰、 提高测量过程仪器的稳定性与测量结果的准确性, 众多研究者采用惰性气体(如: 氮气、 氩气等)排除紫外分光光度计内部的氧气[10, 11], 实现紫外区“ 无氧” [12, 13]。 相比于氩气, 氮气是一种经济、 环保、 易得的气体, 并且氮气只有吸收波长< 79.6 nm的光才开始离解, 在深紫外区中的峰值吸收仅为10-21 cm2-[14], 其大小可以忽略不计。 氮气可作为深紫外区测量时的理想保护气体[9]。
本研究通过对传统的紫外分光光度计加装氮气输配系统, 设计研制了一台深紫外光谱仪, 研究了氮气输配系统中的氮气流量参数对深紫外光谱的稳定性影响, 并且设计了自动进样流通池及进样托盘, 实现了装置密闭条件下自动进样功能, 避免换样过程开关盖板导致氮气外泄。 以常规工业分析产品之一的
称取一定量的分析纯浓硫酸(98.0%, 国药集团化学试剂有限公司, 中国)配制成
配制0~0.21 g· L-1范围内不同浓度的
如图1(a)所示, DLC55-1附件允许测量的最大光程为b=50 mm, 进样方式沿用传统开关盖板方式。 为避免换样过程开合罩体盖板导致罩体氮气外泄损耗, 基于仪器DLC55-1附件和步进电机控制系统, 设计了进样流通池和进样托盘, 实现了自动进样功能, 如图1(b)所示。 进样托盘(109 mm× 110 mm× 40 mm)可同时容纳多个不同规格流通池[图1(c), 可选规格包括1, 5, 10, 20, 50, 80, 100 mm光程], 便于在测量间隙根据样品浓度选取合适光程的流通池。 当流通池规格不满足样品浓度上限要求时, 检测结果将超出量程上限, 导致样品浓度无法被准确定量; 当流通池规格远远高于样品浓度上限要求时, 检测过程的相对误差将显著增大, 导致检测的准确度下降。 进样托盘两侧分别开有多个通光孔(13 mm× 20 mm)[图1(d)], 放置的流通池应与通光孔一一对应。 如图1(e), 传动装置的两端分别与进样托盘和滑道相连, 将进样托盘悬置于底座上方。 此外, 如图1(b)和(d)所示, 流通池的下方和上方分别接有进液管路和出液管路。 检测过程中, 蠕动泵正转驱动进液管路, 使待测样品进入并充满流通池(红色箭头)。
 | 图1 自动进样流通池、 自动进样装置构件图 (a): DLC55-1附件实物图; (b): 进样流通池和进样托盘实物图; (c): 不同规格流通池三维效果图; (d): 进样流通池和进样托盘三维效果图; (e): 步进电机、 传动机构实物图Fig.1 Component diagram device for automatic injection of flow cell and sample introduction (a): Physical picture of the DLC55-1 accessory; (b): Physical picture of the flow cell and quintuplet tray for sample introduction; (c): 3D effect diagram of flow cells with different specifications; (d): 3D effect diagram of the flow cell and quintuplet tray for sample introduction; (e): Physical picture of the stepping motor and transmission mechanism |
采用CMOS高速摄像机拍摄了10, 30, 50, 70, 90和100 r· min-1不同进样速度下
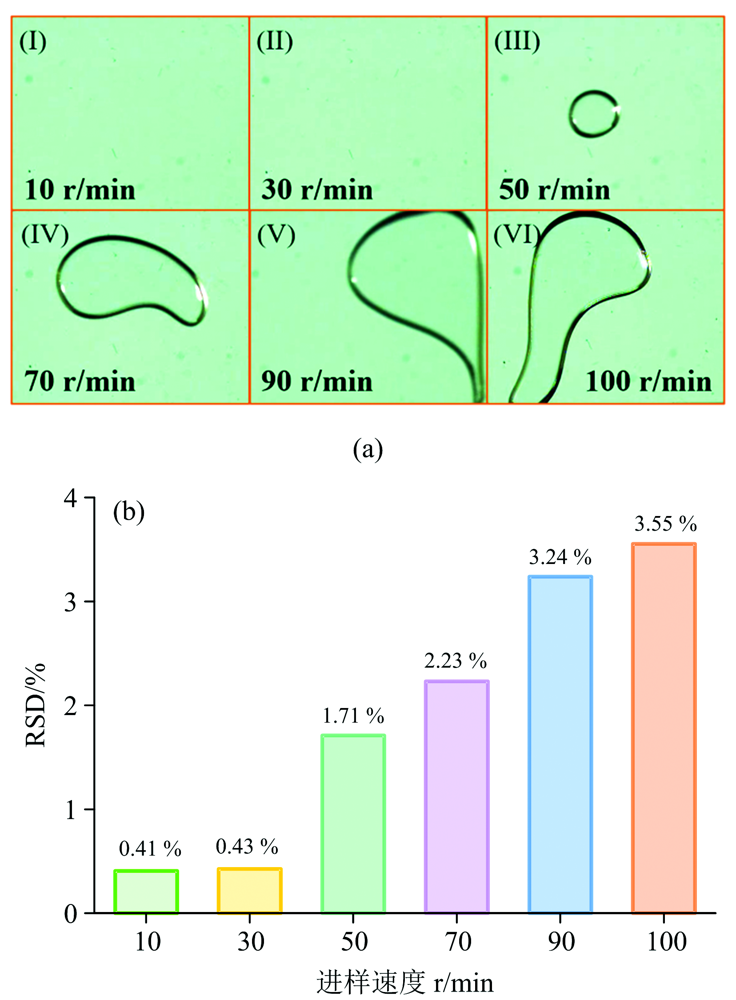 | 图2 (a) 10~100 r· min-1不同进样速度下 |
为隔绝灰尘和环境中温度及湿度变化等因素对仪器运行的干扰, 同时稳定氮气气氛, 在仪器外部搭设密闭罩体以保证氮气在仪器内循环使用, 如图3(a)所示。 罩体底板的四角处设有4个罩体进气口(ϕ =5 mm), 由1个质量流量控制器控制通入罩体内部的氮气流量。 罩体底板还设有3个腔室进气口, 分别由3个质量流量控制器控制通入光学系统区、 样品室和数据接收区的氮气流量。
 | 图3 装置三维效果图和构件实物图Fig.3 3D diagram of the installation and physical drawing of the components |
氮气输配系统其余构件如下: 减压阀[0870301, GCE Market(全球资本设备市场公司, 美国], 过滤器组件[015QPS, 海诺斯(漳州)工业机械有限公司, 中国], 质量流量控制器[S48-32/HMT, 厚礼博精密仪器(北京)有限公司, 中国], 固定式气体检测仪(检测氧气浓度, 同步显示温度及湿度, 威海精讯畅通电子科技有限公司, JXBS-4001), 降温冷水机(HS-28A, 广东海利集团有限公司, 中国, 配合使用ST30散热器, Alphacool公司, 德国), 压差仪(2000-100PA, Dywer仪器仪表制造有限公司, 美国)。 蠕动泵(BT100-2J, 配备探头型号: YZ1515x, 软管型号: 25#, 管内径: 4.8 mm, 保定兰格恒流泵有限公司, 中国)。 加装氮气输配系统的紫外分光光度计设备三维效果图如图3(b)所示。
仪器使用前预热15 min, 开启氮气钢瓶, 待装置内部充满氮气气氛并且仪器条件满足检测要求后采用光谱扫描模式以慢速、 1 nm的扫描间隔连续扫描180~360 nm波长范围, 光谱带宽设置成2 nm, 以获得待测样品的光谱数据。
试验选用不同尺寸的石英材质流通池: b=1 mm, b=5 mm, b=10 mm, b=20 mm, b=50 mm, b=80 mm, b=100 mm。 选择超纯水为基底溶液, 依次采用不同尺寸流通池测定不同浓度的
研究在紫外分光光度计外部设置罩体以有效隔绝氧气进入仪器。 如果氮气通入罩体和仪器内的流量一致时, 过大的氮气流量可能导致仪器内部光学元器件振动而引起仪器检测精度降低, 而过小的氮气流量则导致氮气置换氧气的时间大幅度延长。 因此, 为提高仪器的稳定性, 需对通入仪器内部的氮气流量进行精准控制。
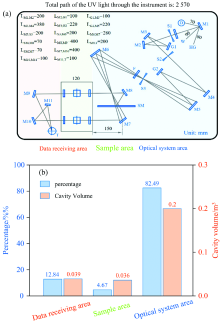
紫外分光光度计内部区域可按功能大致分为光学系统区、 样品室和数据接收区等腔室。 在光学系统区[图4(a)], 氘灯光源发出的复合光线先后经准直镜, 入射狭缝, 光栅, 出射狭缝, 单色光器等光学元器件以过滤光束中的杂散光。 在样品室中, 当紫外光在透过样品室内的待测样品时, 待测样品会吸收特定波长下的紫外光能量, 使紫外光线强度衰减。 在数据接收区, 检测器将接收透过待测样品的紫外光的能量并将接收到的光信号转化成电信号并输出。 显然, 紫外光线在三个腔室内因氧气吸收而出现的光强度衰减程度取决于紫外光线在该腔室内所需穿行的路程。 如图4(b)所示, 紫外光线在光学系统区内所需穿行的路程最长(约2 120 mm), 占总路程比最高(82.49%), 这是因为紫外光线需在光学系统区内经过若干次的反射和折射以滤去杂散光, 相应光学系统区的体积也最大(0.02 m3)。 紫外光线在样品室和接收区内所需穿行的路程则分别为120和330 mm, 占总路程比分别为4.67%和12.84%, 样品室和数据接收区腔室体积分别为0.036和0.039 m3。 由此可见, 紫外光线在三个腔室内经过的光线路程和三个腔室体积存在明显差异, 因此需要分别调控氮气通入不同腔室的流量。
 | 图4 紫外分光光度计仪器内部构件参数图 (a): 光线由光源发出至接收器接收总路程示意图; (b) 光线经过三个腔室的路程占总路程比及三个腔室的体积占总体积比D: 氘灯光源; W: 钨灯光源; HG: 汞灯光源; M1— M11: 准直镜; G1: 一级光栅; G2: 二级光栅; S1: 入射狭缝; S2: 出/入射狭缝; S3: 出射狭缝; F: 滤光片; SM: 斩光器; T: 接收器Fig.4 Parameter diagram of the internal components of the ultraviolet visible spectrophotometer (a)Schematic diagram of the total distance from the light source to the detector; (b) The proportion of the distance of the light passing through the three chambers to the total distance and the volume of the three chambers to the total volume D: Deuterium lamp light source; W: Tungsten lamp light source; HG: Hg-lamp light source; M1— M11: Collimating Mirror; G1: First-order grating; G2: Second-order grating; S1: Entrance slit; S2: Entrance/Exit slit; S3: Exit slit; SM: Chopper; T: Detector |
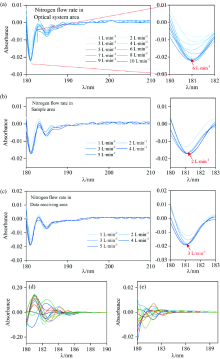
在调控氮气流量时, 以空气为参比测定氮气条件下的吸光度, 即不放置样品, 此时吸光度检测值仅源于紫外光线因环境吸收而出现的光强度衰减。 如图5所示, 以空气为参比时, 不同氮气流量下的吸光度检测值为负。 换而言之, 相比较空气条件, 氮气条件下的紫外光线在传播时的光强度衰减更小。 图5(a)中, 当通入光学系统区的氮气流量数值从1 L· min-1逐渐增大至6 L· min-1时, 在相同的检测时间范围内, 以空气为参比的吸光度检测值逐渐降低。 当氮气流量进一步增加至10 L· min-1后, 吸光度检测值趋于稳定。 吸光度检测值随氮气流量(1~6 L· min-1)的增大而减小的原因显然是较低的氮气流量(< 6 L· min-1)不足以在有限的时间内完全置换出光学系统区内部空气。 此时在腔室内传播的紫外光线将被腔室内部残留氧气吸收, 紫外光线的强度出现不同程度的衰减。 当氮气流量增大至6 L· min-1时, 向腔室内通入的氮气能够在较大程度上隔绝腔室内部氧气, 将紫外光线受氧气吸收强度降至最低。 当氮气流量进一步增大(7, 8, 9和10 L· min-1)时, 吸光度检测值趋于稳定, 意味着在该流量下氮气能够完全隔绝腔室内氧气。 考虑到过高的氮气流量既增大了氮气消耗量并干扰光学元器件的运行, 因此控制通入光学系统区的氮气流量为6 L· min-1。 样品室和数据接收区腔体内不同氮气流量光谱曲线如图5(b)和(c)所示: 同图5(a)的分析, 通入样品室和数据接收区的最佳氮气流量分别为2和3 L· min-1。
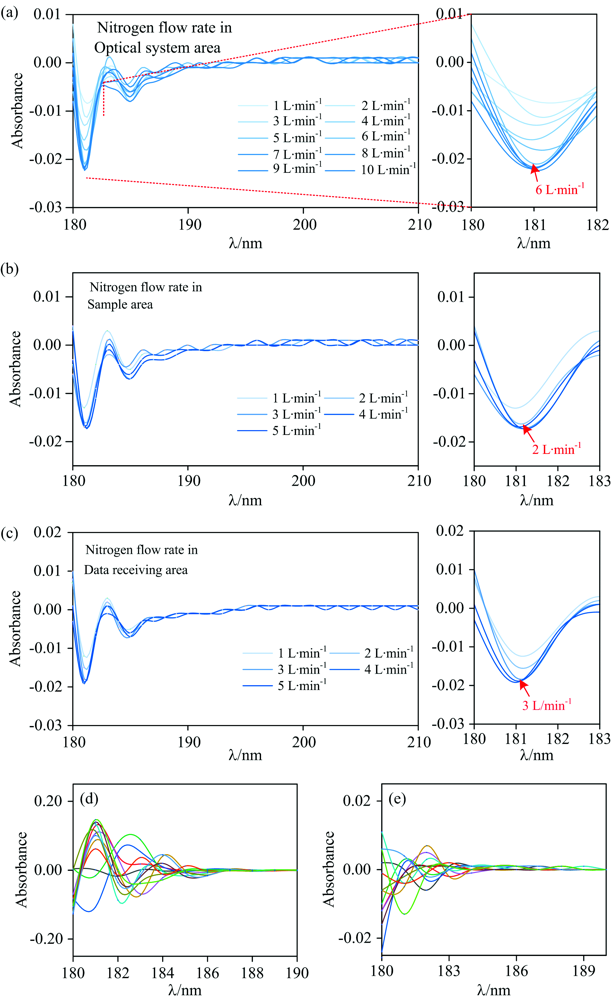 | 图5 (a)光学系统区, (b)样品室内, (c)数据接收区内不同氮气流量的光谱曲线图以及(d)空气和(e)氮气气氛下的基线平直度光谱曲线图(两种气氛下均连续、 慢速扫描12次)Fig.5 Spectral profiles of (a) optical system area, (b) sample area, (c) data receiving area with different nitrogen flow rates and baseline flatness spectral profiles in (d) air and (e) nitrogen atmosphere (12 consecutive, slow scans in both atmospheres) |
通过基线平直度对比验证最佳氮气流量参数下仪器检测结果稳定性。 如图5(d)和(e)所示, 在波长范围为180~190 nm、 波长扫描间隔为1 nm、 扫描速度为慢速的参数设置下连续扫描12次, 得到两种气氛下的基线平直度光谱曲线。 氮气气氛下12次扫描结果的基线平直度平均值为0.010, 相比较空气气氛下的基线平直度平均值(0.108)衰减了90.7%。
图5(d)和(e)中基线平直度的显著差异明显归因于氮气气氛抑制了氧气对紫外线的吸收, 显著降低了光谱曲线的光度噪声, 有效地提高了检测系统的稳定性。
紫外分光光度法的原理是基于样品对紫外光能量的吸收对样品进行定量分析。 由于检测过程中还存在环境吸收而导致的紫外光衰减, 紫外分光光度法的灵敏度和样品吸光度之间存在相互关系。 为探究氮气保护气氛对紫外分光光度法的吸光度及灵敏度的影响, 在不同光程、 不同样品浓度下对比空气和氮气气氛下的检测结果差异。 H2SO4是常用的化学分析产品之一, H2SO4在水溶液中能够完全电离出H+和
设置光程长度为1 mm, 在空气和氮气气氛下分别获得不同浓度下的
 | 图6 b=1 mm时不同浓度 |
从图6(a)和(c)可以看出, 光程为1 mm时, 空气气氛下曲线的最大吸收波长位于184 nm处, 氮气气氛下最大吸收波长位于182 nm处。 普朗克公式揭示光谱曲线波长与样品吸收能量成反比例[16], 因此, 图6(a)和(c)中最大吸收波长的蓝移表明氧气隔绝状态下
如图6(b)和(d)可知, 在一定
为探究氮气气氛对不同光程下
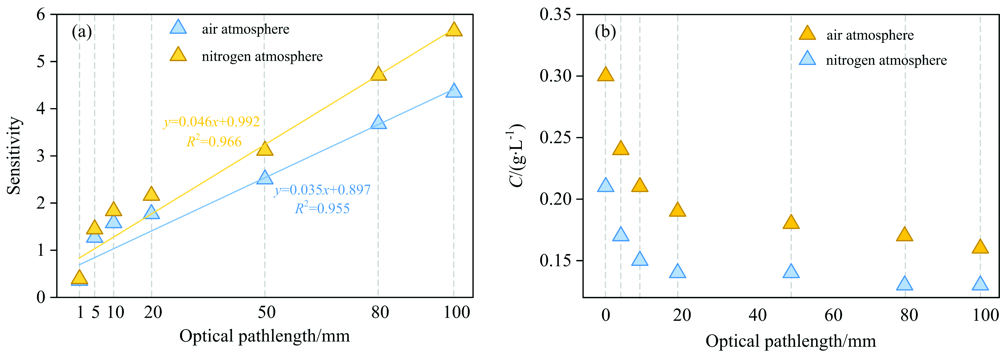 | 图7 空气和氮气气氛下溶液中 |
| 表1 不同光程下的灵敏度及灵敏度变化量和线性范围变化量汇总 Table 1 Summary of sensitivity, sensitivity variation, and linear range variation under different optical path lengths |
离子色谱法是测量
| 表2 离子色谱法和紫外分光光度法测定七种不同浓度 |
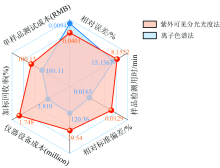
 | 图8 本研究方法(橙色)与离子色谱方法(蓝色)综合性能对比图Fig.8 Comparison diagram of comprehensive performance between this research method (orange) and the ion chromatography method (blue) |
从图8中的数据可以看出, 离子色谱方法(蓝色)与本研究方法(橙色)的检测结果的
为了提高紫外分光光度法的检测结果准确度, 本研究设计并优化了紫外分光光度计的氮气输配系统并对深紫外光谱的稳定性、 吸光度和灵敏度进行了研究。 同时设计了自动进样流通池及进样托盘, 以实现系统密闭情况下的自动进样功能。 通过针对性地精准调控仪器内部光学系统区、 样品室和数据接收区的氮气流量(分别为6, 2和3 L· min-1), 使基线平直度平均值降至0.010, 远低于空气气氛下的基线平直度平均值(0.108), 此时仪器具有良好的稳定性。 确保氮气充满装置后, 调控氮气以微流量(0.6 L· min-1)在腔室内外循环流通, 腔室内充满氮气的同时减少检测过程的氮气消耗量。 以常规工业分析产品之一的
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|