

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Au聚体的吸附位点对杀草强分子表面增强拉曼光谱的影响研究
[顾一帆1  , 廉帅
, 廉帅1 , 高勋1, * , 宋少忠2, * , 林景全1 ]
, 廉帅, 宋少忠, 林景全|
|


作者简介: 顾一帆, 1995年生, 长春理工大学物理学院硕士研究生 e-mail: 864434118@qq.com
杀草强是一种白色结晶粉末状的化学除草剂, 对环境有极强的破坏性, 大量使用会造成农残污染, 对生物体具有致癌作用。 目前利用密度泛函理论探究杀草强分子的拉曼增强机理的相关研究相对较少, 开展了Au聚体吸附位点对杀草强分子表面增强拉曼光谱的影响研究。 采用Multiwfn软件结合VMD软件探究了杀草强分子表面静电势分布, 得出N1, N4和N6是杀草强分子与Au原子配位的最佳位置。 基于密度泛函理论, 运用GaussView5.0和Gaussian09软件, 在B3LYP/6-31++G(d, p)基组水平上对杀草强分子进行几何构型优化, 并对C, H, N原子使用6-31++G(d, p)基组, Au原子使用LANL2DZ赝式基组, 计算了杀草强分子的常规拉曼散射光谱和杀草强分子与Au4聚体以及Au6聚体吸附的表面增强拉曼散射光谱, 并进行特征峰指认和比较。 结果发现在Au与N1配位形成的复合物中, 在1 064, 1 200, 1 392和1 592 cm-1处杀草强分子的拉曼活性明显; Au与N4配位形成的复合物中, 在1 304 cm-1处杀草强分子的拉曼活性明显; Au与N6配位形成的复合物中, 在1 064, 1 520和1 592 cm-1处杀草强分子的拉曼活性明显。 经过比较得出Au与N1和N6配位形成的复合物增强效果较好。 Au4聚体以及Au6聚体与N1吸附得到拉曼特征峰最大增强因子分别达到41和81倍; Au4聚体以及Au6聚体与N6吸附得到拉曼特征峰最大增强因子分别达到55和96倍。 杀草强分子与Au原子结合有很明显的拉曼增强效应, 当Au聚体由四个增加到六个时, Raman光谱增强效果明显。 该研究结果为杀草强分子的实验研究打下了理论基础。
Amitrole is a chemical herbicide in the form of white crystalline powder.It is extremely destructive to the environment, and heavy use can contaminate food and cause cancer. There are relatively few researches on Raman enhancement mechanism of amitrole molecules using density functional theory. Therefore, a model describing the adsorption pattern between the molecule and the substrate was established to predict the surface-enhanced Raman spectrum of amitrole molecules. First, Multiwfn and VMD software were used to calculate the surface electrostatic potential distribution of amitrole molecules and search for the best coordination position between amitrole molecules and Au atoms. It can be obtained that N1, N4 and N6 is the best place for amitrole molecules to coordinate with Au. Based on density functional theory, software GaussView5.0 and Gaussian09, was used to optimize the geometric configuration of amitrole molecules by B3LYP/6-31++G(d, p) basis set. And 6-31++G(d, p)(C, H, N)/Lanladz(Au) basis set was used to calculate the conventional Raman scattering spectra of amitrole molecules and the surface adsorption of amitrole molecules and Au4 clusters and Au6 clusters enhanced Raman scattering spectra. Finally, feature peak identification and comparison are carried out. The results show that the position of the characteristic peaks does not change greatly, but the intensity of some characteristic peaks increases obviously. In the complex formed by coordination between Au and N1, the Raman activity of amitrole molecules was obvious at 1 064, 1 200, 1 392 and 1 592 cm-1. In the complex formed by coordination between Au and N4, the Raman activity of amitrole molecules was obvious at 1 304 cm-1. In the complex formed by coordination between Au and N6, the Raman activity of amitrole molecules was obvious at 1 064, 1 520 and 1 592 cm-1. Through comparison, the compound formed by coordination between Au and N1, N6 has a better enhancement effect, and the maximum enhancement of characteristic peak reached 41, 81, 55 and 96 times respectively. The results show that the complexes formed by coordinating Au with N1 and N6 have a better reinforcement effect. The maximum enhancement factor of Au4 polymer and Au6 polymer with N1 adsorption reached 41 and 81 times respectively; The maximum enhancement factor of Au4 polymer and Au6 polymer with N6 adsorption reached 55 and 96 times respectively. Amitrole molecules and Au atoms have an obvious Raman enhancement effect. When the number of Au polymers increases from four to six, the Raman spectrum enhancement effect is obvious. This work laid a foundation for studying the SERS enhanced mechanism of amitrole molecules.
杀草强(C2H4N4), 又称为3-氨基-1, 2, 4-三氮唑, 它是一种白色结晶状粉末, 常用作于化学除草剂, 可防止多年内生出杂草。 但是杀草强的使用也存在一些隐患, 研究表明, 杀草强能通过植物[1]和水介质[2]造成食物污染, 经小鼠、 大鼠口服及皮下注射会发生甲状腺及肝脏肿瘤, 有一定的潜在致癌作用[3]。 目前人们常用的检测方法有高效液相色谱-串联质谱法[4, 5], 而此方法具有操作繁琐、 样品制备复杂、 对样品有损害等缺点。 表面增强拉曼光谱技术(surface-enhanced Raman spectroscopy, SERS)由于具有高灵敏度的优点, 能够检测吸附在金、 银溶胶等表面的待测物分子, 使待测物信号比普通拉曼信号增强104~106倍[6, 7]。 但是, 随着基底性质的改变, 吸附分子的Raman光谱也将改变, 这对研究带来了困难。 因此, 可以通过建立一个能描述分子和基底之间吸附形式的模型, 用来预测分子的表面增强拉曼光谱[8]。 密度泛函理论方法(density functional theory, DFT)结合拉曼光谱方法共同研究物质已经成为当前的一种有效的研究手段。 Ismail Abdulazeez等[9]运用DFT计算了普鲁卡因分子的SERS光谱, 增强来源于普鲁卡因分子中的N原子与Ag表面相互作用。 Nandita Maiti等[10]指出氨基聚羧酸分子与金属原子结合至少有两种可能的结合方式, DFT预测表明氨基聚羧酸分子通过N原子和O原子直接吸附到Ag表面, 使得拉曼信号增强。 陈玉锋等[11]运用DFT理论计算了杀草强分子的Raman光谱并且分析了杀草强和Ag可能吸附的位置。 本文从理论计算方面对杀草强分子的SERS谱进行了研究, 通过对杀草强分子静电势的分析, 寻找了该分子与Au原子配位的合适位置。 在此基础上分别对杀草强分子与4个Au配位的复合物(4Au-C2H4N4)和6个Au配位的复合物(6Au-C2H4N4)的SERS谱进行分析, 为研究杀草强分子的SERS增强机理打下了基础。
采用Multiwfn结合VMD软件计算杀草强分子表面静电势分布[12], 寻找杀草强分子与Au原子配位的最佳位置。 基于DFT理论, 使用GaussView5.0和Gaussian09软件, 在B3LYP/6-31++G(d, p)基组水平上对分子结构优化, 并对杀草强分子分别配位4个Au原子和6个Au原子形成的复合物采用B3LYP方法, 对C, H, N原子使用6-31++G(d, p)基组, Au原子使用LANL2DZ赝式基组, 进行结构优化与振动频率计算。 杀草强分子的振动模式通过VEDA4软件进行分析, 在DELL工作站上进行计算。
杀草强分子式为C2H4N4, 由C, H和N元素组成, 分子量为84.08, 是一种白色结晶粉末, 易溶于水。 基于DFT理论, 在B3LYP/6-31++G(d, p)基组水平上对分子结构优化并进行振动频率计算, 所得振动频率均大于0, 未发现虚频存在, 计算得到的C2H4N4分子结构图与陈玉锋等[11]计算的结果一致。
分子静电势是判断分子反应活性和识别分子的有效途径[13], 运用Multiwfn软件结合VMD软件计算得到杀草强分子的静电势如图1所示。 红色区域表示正电荷或亲核区域, 亲核试剂倾向于和电正性物种结合; 蓝色区域则表示负电荷或亲电区域, 亲电试剂由于缺少电子, 容易进攻反应物上带负电荷的位置。 而金属离子都是属于亲电试剂, 由于其缺少电子, 容易发生亲电反应。 从杀草强的分子表面静电势图中可以看出, 若想要使金属Au与杀草强分子配位, 理论上Au应该选择蓝色区域位置的N原子。 由于N原子在形成化合物作为中心时, 常保留有孤电子对, 因此这样的化合物可作为电子对给予体, 向金属离子配位。 因此, Au聚体最有可能和杀草强分子中的N原子吸附增强。
 | 图1 杀草强分子的表面静电势图Fig.1 Surface electrostatic potential of Amitrole |
通过Multiwfn软件计算出杀草强分子中每个原子的静电势统计数据, 如表1所示。 由表1中的负值区域的静电势平均值可以看出, 氮原子(1号)、 氮原子(4号)和氮原子(6号)附近的分子表面静电势平均值分别为-21.274, -23.504和-23.846 kcal· mol-1。 由于其附近分子表面没有正值区域, 正值区域的静电势平均值在Multiwfn软件中显示的是NaN。 杀草强分子在N1, N4和N6原子上的电子密度较高, 电负性较强, 因此杀草强分子与Au配位主要是通过N1, N4和N6原子连接形成增强的。 陈玉锋等[11]通过分子静电势图分析得出杀草强分子和金属SERS基底主要通过N1和N4原子发生作用, 而本文通过对杀草强分子表面静电势图进行分析, 结合计算出的杀草强分子静电势平均值, 比较得出Au纳米颗粒都有可能与N1, N4和N6原子配位形成新的配合物。
| 表1 杀草强分子静电势平均值 Table 1 Average electrostatic potential of Amitrole |
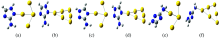
优化后的杀草强分子与Au纳米颗粒配位体系的分子结构如图2所示。 计算结果中无虚频, 表明优化后的4Au-C2H4N4, 6Au-C2H4N4分子是稳定的。 对于N配位Au后, 由表2杀草强分子的结构参数, 可以看到键长和含C和N的杂环化合物的夹角发生细微变化。
 | 图2 杀草强金配合物分子结构图 (a): 4Au-C2H4N4(N1); (b): 6Au-C2H4N4(N1); (c): 4Au-C2H4N4(N4); (d): 6Au-C2H4N4(N4); (e): 4Au-C2H4N4(N6); (f): 6Au-C2H4N4(N6)Fig.2 Amitrole gold complex molecular structure diagram (a): 4Au-C2H4N4(N1); (b): 6Au-C2H4N4(N1); (c): 4Au-C2H4N4(N4); (d): 6Au-C2H4N4(N4); (e): 4Au-C2H4N4(N6); (f): 6Au-C2H4N4(N6) |
| 表2 杀草强分子结构参数 Table 2 The structure parameter of Amitrole |
在图2中随着Au原子的增加, 4Au-C2H4N4(N1)和6Au-C2H4N4(N1)的N2-N1-C5的夹角分别为103.676° 和103.145° , N原子与Au原子之间的距离由2.106 Å 增大到2.184 Å , 四个Au原子组成的二面角为179.848° , 几乎处于同一平面, 两个Au原子与N1及相邻的N2所形成的二面角为91.715° , 说明Au所构成的平面与含C和N的杂环化合物几乎构成面面垂直。 随着Au原子的增加, 4Au-C2H4N4(N4)和6Au-C2H4N4(N4)的C3-N4-C5的夹角分别为105.504° 和105.127° , N原子与Au原子之间的距离由2.113 Å 增大到2.186 Å , 四个Au原子组成的二面角为179.415° , 几乎处于同一平面, 两个Au原子与N4及相邻的C3所形成的二面角为91.636° , 说明Au所构成的平面与含C和N的杂环化合物几乎构成面面垂直。 随着Au原子的增加, 4Au-C2H4N4(N6)和6Au-C2H4N4(N6)的N1-C5-N4的夹角分别为115.706° 和115.507° , N原子与Au原子之间的距离由2.248 Å 增大到2.342 Å , 四个Au原子组成的二面角为179.940° , 几乎处于同一平面, Au原子与氨基所构成的二面角为109.955° , 说明Au原子所构成的平面与氨基几乎构成面面垂直。 结果表明当金配合物由四个增加到六个时, Au与N之间的键长变大, 含C和N的杂环化合物的夹角变小, 杀草强分子与Au纳米颗粒配位体系形成的原子聚体变紧密。
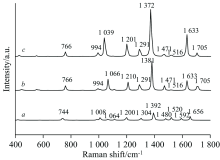
图3、 图4、 图5分别是N1, N4和N6原子与Au原子配位的杀草强分子的SERS光谱。 从图3中可以看出, 加入金纳米原子后, 相对于杀草强分子C2H4N4的Raman光谱, 4Au-C2H4N4(N1)和6Au-C2H4N4(N1)两个分子体系的Raman光谱中的Raman特征峰存在一定的红移, 且Raman特征峰强度有明显增强。 在C2H4N4分子中, Raman谱峰1 064 cm-1对应了N— N伸缩振动、 H— N— C面内弯曲振动和N— C— N面内弯曲振动, Raman光谱强度较弱; 在C2H4N4-Au4中谱线蓝移至1 066 cm-1处, 且由一弱峰变成了强峰光谱增强了41倍; 在C2H4N4-Au6中红移至1 039 cm-1处, Raman光谱强度继续增强, 增强了81倍。 说明在吸附到金纳米颗粒表面后, 由于N— N伸缩振动、 H— N— C面内弯曲振动和N— C— N面内弯曲振动使得1 064 cm-1处的特征峰变成了一个强峰。 这些显著的变化也证明了与Au相连接的原子附近局域环境发生了较大程度的变化。 Raman谱峰1 200 cm-1对应了C2H4N4分子中N— C伸缩振动和H— N— C面内弯曲振动, 光谱强度较弱, 在C2H4N4-Au4中蓝移至1 210 cm-1处, 且Raman光谱强度增强了14倍; 在C2H4N4-Au6中蓝移至1 201 cm-1处, Raman光谱强度增强了28倍。 Raman谱峰1 392 cm-1对应了C2H4N4分子中N— C伸缩振动和H— N— C面内弯曲振动, Raman光谱强度较弱; 在C2H4N4-Au4中红移至1 381 cm-1处, Raman光谱强度增强了8倍; 在C2H4N4-Au6中红移至1 372 cm-1处, Raman光谱强度增强了15倍。 Raman谱峰1 592 cm-1对应了C2H4N4分子中N— C伸缩振动、 H— N— C面内弯曲振动和H— N— H面内弯曲振动, 在C2H4N4-Au4中蓝移至1 633 cm-1处, Raman光谱强度增强了22倍; 在C2H4N4-Au6中蓝移至1 633 cm-1处, Raman光谱强度增强了47倍。
 | 图3 C2H4N4(N1)的表面增强拉曼光谱图 (a): C2H4N4的拉曼光谱; (b): 4Au-C2H4N4(N1) 的拉曼光谱; (c): 6Au-C2H4N4(N1)的拉曼光谱Fig.3 Surface enhanced Raman spectroscopy of C2H4N4(N1) (a): Raman spectra of C2H4N4; (b): Raman spectra of 4Au-C2H4N4(N1); (c): Raman spectra of 6Au-C2H4N4(N1) |
 | 图4 C2H4N4(N4)的表面增强拉曼光谱图 (a): C2H4N4的拉曼光谱; (b): 4Au-C2H4N4(N4)的拉曼光谱; (c): 6Au-C2H4N4(N4)的拉曼光谱Fig.4 Surface enhanced Raman spectroscopy of C2H4N4(N4) (a): Raman spectra of C2H4N4; (b): Raman spectra of 4Au-C2H4N4(N4); (c): Raman spectra of 6Au-C2H4N4(N4) |
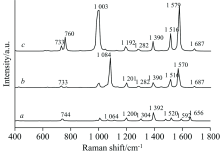
 | 图5 C2H4N4(N6)的表面增强拉曼光谱图 (a): C2H4N4的拉曼光谱; (b): 4Au-C2H4N4(N6)的拉曼光谱; (c): 6Au-C2H4N4(N6) 的拉曼光谱Fig.5 Surface enhanced Raman spectroscopy of C2H4N4(N6) (a): Raman spectra of C2H4N4; (b): Raman spectra of 4Au-C2H4N4(N6); (c): Raman spectra of 6Au-C2H4N4(N6) |
从图4中可以看出, 加入金纳米原子后, 相对于杀草强分子C2H4N4的Raman光谱, 4Au-C2H4N4(N4)和6Au-C2H4N4(N4)两个分子体系的Raman光谱中只有在1 304 cm-1处存在红移现象, 且发生了明显的Raman光谱增强。 在4Au-C2H4N4(N4)和6Au-C2H4N4(N4)两个分子体系1 304 cm-1处Raman光谱红移至1 300和1 291 cm-1处, 并且Raman光谱强度分别增强了20和74倍。
当Au纳米颗粒与杀草强C2H4N4分子中的N6结合配位时, 相对于杀草强分子C2H4N4的Raman光谱, 4Au-C2H4N4(N6)和6Au-C2H4N4(N6)两个分子体系的Raman光谱中的Raman特征峰存在一定的红移, 且Raman特征峰强度有明显增强。 在C2H4N4分子中, Raman谱峰1 064 cm-1对应了N— N伸缩振动、 H— N— C面内弯曲振动和N— C— N面内弯曲振动, 对应一较弱的Raman谱峰; 在C2H4N4-Au4中蓝移至1 084 cm-1处, 且出现了较强的光谱增强, Raman光谱强度增强了55倍; 在C2H4N4-Au6中红移至1 003 cm-1处, Raman光谱强度增强了96倍。 在C2H4N4分子中, Raman谱峰1 520 cm-1对应了N— C伸缩振动和H— N— N面内弯曲振动, 对应一较弱的Raman谱峰; 在C2H4N4-Au4中红移至1 516 cm-1处, Raman光谱强度增强了13倍; 在C2H4N4-Au6中红移至1 516 cm-1处, Raman光谱强度增强了27倍。 在C2H4N4分子中, Raman谱峰1 592 cm-1对应了N— C伸缩振动、 H— N— C面内弯曲振动和H— N— H面内弯曲振动, 对应一较弱的Raman谱峰; 在C2H4N4-Au4中红移至1 570 cm-1处, Raman光谱强度增强了25倍; 在C2H4N4-Au6中红移至1 579 cm-1处, Raman光谱强度增强了55倍。
Au原子的接入增强了N— N、 N— C之间的伸缩振动和H— N— C面内弯曲振动、 H— N— H面内弯曲振动, 整个Raman光谱的增强效果集中在700~1 700 cm-1区域。 同时由表2杀草强分子结构参数可以看出, 由于配位了Au聚体, 距离Au较近的N原子振动强烈, 键长变长, 键角变小, 原子聚体变紧密, 因此导致了Raman特征峰的频移和强度变化。 通过对比分析, 发现杀草强分子在N1和N6上连接Au原子的Raman增强程度最高。 Au与N1配位形成的4Au-C2H4N4和6Au-C2H4N4复合物特征峰最大增强达到41倍和81倍; Au与N6配位形成的4Au-C2H4N4和6Au-C2H4N4复合物特征峰最大增强达到55倍和96倍, 从而得出N1和N6是最佳的金结合位点。 根据表面选择规则[14], 可以得出C5-N1和氨基接近并垂直金表面, 从而导致N和金锚定, 产生电荷转移效应, 分子极化率增大从而使得谱峰选择性增强。
分析了杀草强分子的表面静电势分布, 寻找了杀草强分子与Au原子配位的最佳位置。 运用DFT理论, 采用B3LYP方法, 对杀草强分子中的C, H, N原子使用6-31++G(d, p)基组, Au原子使用LANL2DZ赝式基组。 计算结果表明, 4Au-C2H4N4和6Au-C2H4N4两个分子体系中, 在N1和N6上连接Au原子的增强效果比较好, 特征峰最大增强分别达到41、 81倍和55、 96倍, 且Raman谱峰位置发生一定程度的红移或者蓝移。 当Au聚体由四个增加到六个时, 杀草强C2H4N4分子Raman光谱增强效果更显著。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|