



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函理论的维生素C紫外光谱与激发性质的计算
[林艳1  , 苏俊宏
, 苏俊宏1, * , 唐延林2 , 杨丹3 ]
, 苏俊宏, 唐延林|
|


作者简介: 林 艳, 1986年生, 西安工业大学光电工程学院博士研究生 e-mail: 572442382@qq.com
维生素C为酸性己糖衍生物, 有L-型(抗坏血酸(AA))和D-型(脱氢抗坏血酸(DHA))两种异构体, DHA是AA的第一个稳定氧化产物, 是AA的可逆氧化形式, 因此, 对AA的任何性质或度量的讨论都将涉及同一体系中DHA的性质。 紫外光谱是电子跃迁难易程度和几率的直观体现, 理论计算方法与分子模型的构建不合理, 都将导致对维生素C的最大吸收峰产生误判, 从而无法准确的表征维生素C的激发性质。 因此, 为准确探究维生素C的抗氧化机理, 在液相环境中, 基于密度泛函理论(DFT)和含时密度泛函(TD-DFT)理论, 分别采用pbepbe/6-311++g(2d, 2p)方法和B3LYP/6-311++g(2d, 2p)方法, 计算并分析了维生素C的抗坏血酸和脱氢抗坏血酸分子的结构、 紫外光谱及电子激发特征。 结果表明: pbepbe/6-311++g(2d, 2p)是计算AA紫外吸收光谱更精确的方法; DHA比AA的环状结构发生了显著的平面扭曲。 紫外光谱分析可知, 基态跃迁到S1, S2, S3, S4, S14和S18激发态为AA产生紫外光谱的主要原因, AA位于200.171 5 nm处的吸收峰包含 n→ π*, n→ σ*电子跃迁, 266.9248 nm处的吸收峰包含 n→ π*和 π→ π*的跃迁。 基态跃迁到S6, S9, S12, S13, S15, S16, S17, S19和S20激发态为DHA产生紫外光谱的主要原因, DHA的最强吸收峰位于181.024 8 nm处, 具有 n→ σ*和 n→ π*的跃迁特征, 231.346 39 nm处微弱的吸收峰指认为 n→ π*跃迁, 282.466 8 nm处的吸收峰主要对应 n→ π*的跃迁; 通过空穴-电子分布及其衍生量的分析, 可定性地对AA吸收峰起主要作用的7个激发态的特征及对DHA吸收峰起主要作用的9个激发态的特征进行详细的指认。 其中对AA紫外光谱起主要贡献的S4, S13和S14激发态与对DHA紫外光谱起主要贡献的S6, S9, S17和S20激发态电荷转移较明显, 空穴的质心中心和电子质心的中心分离较明显, 可以指认为电荷转移激发, 而其他激发态的电子与空穴分离程度很低, 指认为局域激发。
Vitamin C is an acidic hexose derivative, which has two isomers of L-type (ascorbic acid (AA)) and D-type (dehydroascorbic acid (DHA)). DHA is the first stable oxidation product of AA and is the reversible oxidized form of AA. Therefore, any discussion of the nature and measurement of AA will involve the nature of DHA in the same system. The ultraviolet spectrum is a visual representation of how easy and difficult the electron transition is. Unreasonable theoretical calculation method and molecular model construction will lead to misjudgment of the maximum absorption peak of vitamin C, and thus can not accurately characterize the excitation properties of vitamin C. In order to accurately explore the antioxidant mechanism of vitamin C, based on density functional theory (DFT) and time-dependent density functional theory (TD-DFT), the molecular structure, ultraviolet spectrum and electron excitation characteristics of ascorbic acid (AA) and dehydroascorbic acid (DHA) of vitamin C were calculated and analyzed at the level of pbepbe/6-311++g(2d,2p) and B3LYP/6-311++g(2d,2p) in liquid phase environment in this paper. The results showed that the pbepbe/6-311++g(2d, 2p) method is the more accurate method to calculate the ultraviolet absorption spectrum of AA. Compared with AA, the ring structure of DHA has a significant plane distortion than AA. According to the analysis of the spectral contribution shows that the ground state transition to S1, S2, S3, S4, S14, S18 excited state is the main reason for AA ultraviolet spectrum, the absorption peak of AA at 200.171 5 nm contains the electronic excitations of n→ π* and n→ σ* electronic transitions, the absorption peak at 266.924 8 nm contains n→ π* and π→ π* transitions. The reason for the ultraviolet spectrum of DHA is mainly due to the ground state transition to S6, S9, S12, S13, S15, S16, S17, S19, S20 excited state, the strongest absorption peak of DHA is located at 181.024 8 nm, which has the transition characteristics of n→ σ* and n→ π*. The weak absorption peak at 231.346 39 nm refers to the n→ π* transition, and the absorption peak at 282.466 8 nm mainly corresponds to the n→ π* transition. By analysing the hole-electron distribution and its derivatives, it is possible to qualitatively identify the characteristics of the 7 excited states that play a major role in the AA absorption peak and the 9 excited states that make major contributions in the DHA absorption peak. Among them, the S4, S13, S14 excited states that make major contributions to the AA ultraviolet spectrum and the S6, S9, S17, S20 excited states that make major contributions to the DHA ultraviolet spectrum have obvious charge transfer, the centroid center of the hole and the center of the electron centroid are separated, which can be referred to as the charge transfer excitation, the separation of electrons and holes in other excited states is very small, which can be referred to as local excitation.
维生素C(Vitamin C)为酸性己糖衍生物, 有L-型(抗坏血酸(AA))和D-型(脱氢抗坏血酸(DHA))两种异构体, DHA是AA的第一个稳定氧化产物, 是AA的可逆氧化形式, 因此, 对AA的任何性质或度量的讨论都将涉及同一体系中DHA的性质。 早在1970年, 文献[1]通过实验验证AA水溶液在200~350 nm的紫外光谱是C=C双键π → π * 电子跃迁产生的。 而在理论计算方面, 文献[2]在气相和水相环境中, 用B3LYP方法计算左旋抗坏血酸的紫外光谱吸收峰分别为238和247 nm; 据文献[3]报道, AA的最强吸收峰位于265 nm处。 DHA和AA结构的相似与差异性, 决定了DHA特有的生化活性, 二者吸收光谱的差异是测定维生素C衍生物的基础。 文献[4]依据AA的结构模型, 计算了去氢抗坏血酸的几何构型和振动光谱, 并对其特征吸收峰振动归属进行了详细指认; 文献[3]表明, DHA的最强吸收峰位于185 nm处, 与AA不同的是, DHA在220 nm以上的吸收峰强度很弱。
前期计算的紫外光谱[2]特征吸收峰无法准确的表征维生素C的激发性质。 因此, 为准确的探究维生素C的抗氧化机理, 本文以抗坏血酸和脱氢抗坏血酸为研究对象[5], 提出了一种计算AA和DHA紫外-可见吸收光谱更精确的方法, 并详细分析了AA和DHA的紫外光谱、 电子激发态转移过程及其分子性质, 从而为维生素C的生物活性和药物构象关系的相关实验研究提供有益的理论支撑。
紫外光谱是电子跃迁难易程度和几率的直观体现, 电子跃迁是分子发光的机理的核心。 本文采用空穴-电子分布及其衍生量研究维生素C的电子激发特征, 相关参数D, Sr, H, t的表达式如式(1)—式(4)
式中相关量的含义见文献[6, 7], 根据式(1)—式(4)可定性表征分子激发特征, 从而确定电子跃迁类型及电子转移过程。 本文在液相(水)环境中, 利用Gaussian 09软件包, 密度泛函理论[8](DFT), 分别用pbepbe[9]、 B3LYP[10]方法, 在6-311++g(2d, 2p)基组水平上优化了AA和DHA的几何构型, 频率分析无虚频, 表明优化的结果是稳定构型, 并在同样的基组水平上运用含时密度泛函(TD-DFT)理论[11], 计算了AA和DHA的紫外光谱, 并利用Multiwfn波函数软件包[12]详细分析了AA和DHA的紫外光谱特征和激发态性质。
分别用pbepbe和B3LYP方法, 在6-311++g(2d, 2p)基组水平上优化的AA和DHA几何构型如图1所示, 频率分析无虚频, 表明优化的结构是局域能量极小值的稳定构型。
 | 图1 维生素C的几何构型 (a): 抗坏血酸(AA); (b): 脱氢抗坏血酸(DHA)Fig.1 Geometries of Vitamin C (a): Ascorbic acid (AA); (b): Dehydroascorbic acid (DHA) |
图1表明, AA的羰基和烯二醇基相邻, 使得AA具有特有的酸性和还原性, 而DHA具有三个相邻的羰基, 在一定条件下, AA的C2和C3位上两个相邻的烯醇式羟基极易解离或被氧化, 从而转化为DHA。
维生素C的主要化学键长和二面角如表1所示, 计算得到的结构参数和文献[4]报道的一致。 AA和DHA环状结构的主要二面角参数表明, DHA二面角大小比AA二面角大小偏离平面的幅度更明显, 说明DHA比AA的环状结构发生了显著的平面扭曲, DHA的共轭效应减弱, 稳定性降低。
| 表1 维生素C的主要化学键长和二面角 Table 1 Main bond distances and dihedral angles of Vitamin C |
分别使用pbepbe/6-311++g(2d, 2p)和B3LYP/6-311++g(2d, 2p)基组水平, 计算的AA紫外光谱(图2)和DHA紫外光谱(图3)表明: 使用pbepbe/6-311++g(2d, 2p)基组水平得到的AA紫外光谱最大吸收峰位于266.924 8 nm处, 仅比实验测量值[1, 3]红移约1.924 8 nm; 使用B3LYP/6-311++g(2d, 2p)基组水平得到的DHA紫外光谱值最大吸收峰位于181.024 8 nm处, 与文献[3]报道的实验数据吻合较好, 最大吸收峰蓝移约4 nm。
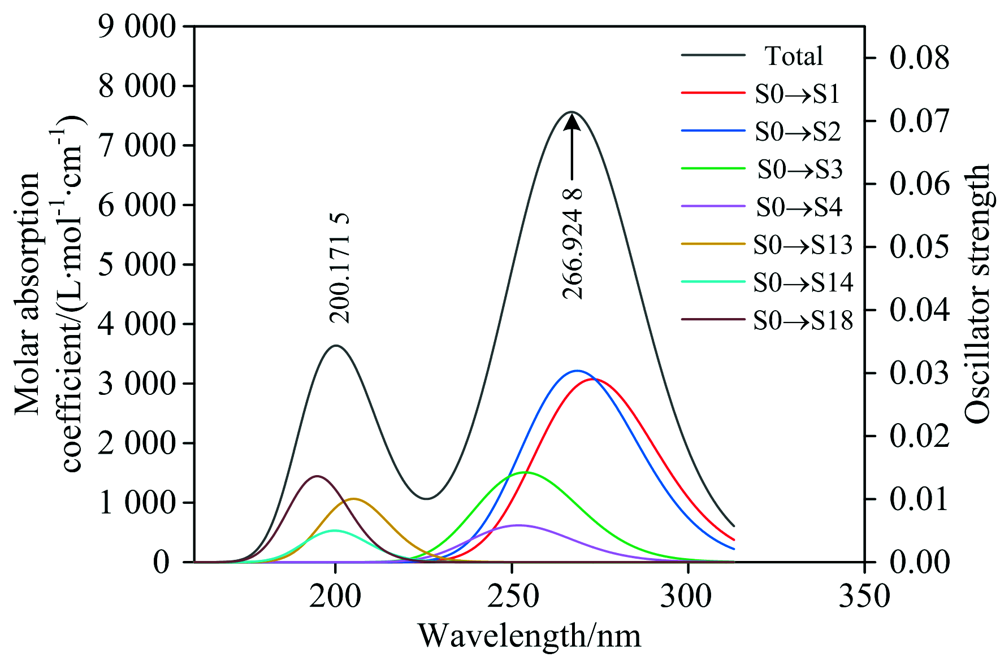 | 图2 AA理论计算的紫外光谱Fig.2 Computational ultraviolet spectrum of AA |
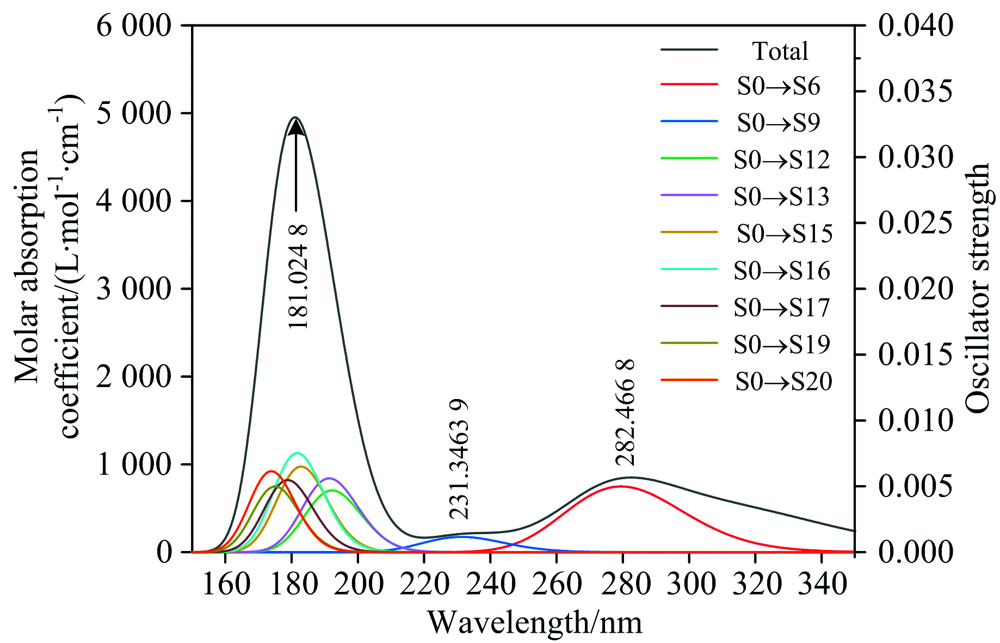 | 图3 DHA理论计算的紫外光谱Fig.3 Computational ultraviolet spectrum of DHA |
图中的黑色曲线(Total)为维生素C各个电子跃迁的吸收曲线归一化之后, 然后再把所有跃迁曲线的Y值相加所得到的总的曲线, 彩色曲线是将振子强度大于某个阈值的跃迁进行高斯函数展宽后得到的曲线, 其中S0表示基态, Sx表示第x个激发态。
图2表明, AA的紫外光谱吸收峰分别位于200.171 5和266.924 8 nm处。 其中, 对200.171 5 nm处吸收峰其主要贡献的激发态包括25%的S0→ S13、 15%的S0→ S14和33%的S0→ S18的激发态, 该吸收峰不仅包含n→ π * 的电子跃迁, 还包含n→ σ * 的电子跃迁; 266.924 8 nm处的最强吸收峰主要包含38%的S0→ S1、 42%的S0→ S2、 14%的S0→ S3和5%的S0→ S4的电子跃迁, 该吸收峰具有n→ π * 和π → π * 的跃迁特征。
图3表明, DHA的紫外吸收峰分别位于181.024 8, 231.346 39和282.466 8 nm处。 其中, 最强吸收峰位于181.024 8 nm处, 主要包含5%的S0→ S12、 7%的S0→ S13、 19%的S0→ S15、 23%的S0→ S16、 16%的S0→ S17、 11%的S0→ S19和11%的S0→ S20的电子跃迁, 该吸收峰对应n→ σ * 和n→ π * 的跃迁特征。
在231.346 39 nm处有一个微弱的吸收峰, 主要对应83%的S0→ S9的跃迁, 归属于n→ π * 跃迁; 282.466 8 nm处的吸收峰主要对应87%的S0→ S6跃迁, 指认为n→ π * 跃迁。
研究[6, 7]表明: D, Sr, H和t是衡量电子激发模式的定量指标, 常被作为指认电子激发类型的重要判据。 计算得到的维生素C主要吸收峰对应激发态的D, Sr, H, t指数如表2所示。
| 表2 维生素C的激发态指数 Table 2 Excited states index of Vitamin C |
如图4、 图5所示, 利用空穴-电子分布图, 空穴、 电子分布的平滑化描述图(Chole-Cele图), 可以直观地考察激发态电子的去向及激发类型。 图中绿色代表电子分布, 蓝色代表空穴分布, 电子跃迁可以认为是从空穴区域跃迁到电子区域。
 | 图4 AA的空穴-电子、 Chole-Cele示意图 第一列和第三列: 空穴-电子图, 第二列和第四列: Chole-Cele图Fig.4 Electron-hole, Chole-Cele distributions of AA The first and third columns: hole-electron distributions; The second and fourth columns: Chole-Cele distributions |
 | 图5 DHA的空穴-电子、 Chole-Cele示意图 第一列和第三列: 空穴-电子图; 第二列和第四列: Chole-Cele图Fig.5 Electron-hole, Chole-Cele distributions of DHA The first and third columns: hole-electron distributions; The second and fourth columns: Chole-Cele distributions |
结合表2和图4可知, AA的D指数较大的是基态到S4, S13和S14的3个激发态, 空穴-电子分布图表明, 基态到S4, S13和S14的电子和空穴分布于不同区域, 空穴和电子分离明显, 因此指认为电荷转移激发; 基态到S1和S2的空穴-电子分布范围在环上有交叉, 空穴和电子分离不明显, 因此, 将基态到S1和S2指认为局域激发。 Chole-Cele分布图表明基态到S4, S13和S14蓝色和绿色等值面的中心(分别对应空穴和电子的质心位置)离得比较远, 而其他激发态的蓝色和绿色等值面中心距离都很近, 只能是局域激发。
Sr指数表明, 激发态1, 2, 3和18的Sr指数相对较大, 由空穴-电子图可知, 主要因为这4种激发在环状结构上的高度局域的π → π * 激发所致。 虽然S0→ S14的激发区域主要发生在环状结构上, 但由于空穴-电子等值面中心距离较远, 表现出n→ σ * 电荷转移激发特征, 因此Sr较小。
结合H指数和空穴-电子图可知: S4和S13激发态的电子跃迁涉及的原子和基团较集中, 使得激发态的电子云往中心聚集得更紧密, 因此H指数较小。 t指数表明, 激发态S4, S13和S14的t指数为正值, 表明空穴和电子分离较为明显, 因此把激发态S4, S13和S14指认为电荷转移激发; 其他t为负值对应的激发态, 说明电子与空穴分离程度很低。
对AA吸收峰起主要作用的7个激发态的特征进行指认如下: 激发态S1, S2和S3: π → π * , n→ π * 局域激发; 激发态S4和S13: n→ π * 电荷转移激发; 激发态S14: n→ σ * 电荷转移激发; 激发态S18: n→ π * 局域激发, 伴随微弱的n→ σ * 局域激发。
同理结合表2和图5, DHA的激发态S6, S9, S17和S20的D指数都较大, 且Chole-Cele图上蓝色等值面(空穴的质心)和绿色等值面(电子的质心)的中心分离较明显, 可以指认为电荷转移激发, 而其他激发态的蓝色和绿色等值面中心距离都很近, 因此指认为局域激发。
Sr指数表明, 激发态S12和S13的Sr指数相对较大, 结合空穴-电子图可知, 这是由于S12和S13激发在环状结构上高度局域的n→ π * 激发所致。 S19的Sr指数偏小, 这是因为S19的电子-空穴分布高度局域在环状结构上, 因此Sr指数重叠的区域也主要集中在环状结构上, 激发主要体现的是n→ σ * 局域激发特征。
结合H指数与空穴-电子图表明: 激发态S6, S9和S17激发态的空穴-电子在原子和基团上的覆盖率不高, 导致这3个激发态的H值相对较小; 而其他激发态的空穴-电子分布覆盖的原子或基团更广, 从而H值较高。 t指数表明, 激发态S6, S9, S17和S20的t指数为正值, 表明空穴和电子分离较为明显, 因此指认为电荷转移激发。
对DHA吸收峰起主要作用的9个激发态的特征进行指认如下: 激发态S6, S9, S17: n→ π * 电荷转移激发; 激发态S12, S13, S15和S16: n→ π * 局域激发; 激发态S19: n→ σ * 局域激发, 并伴随微弱的n→ π * 局域激发; 激发态S20: n→ σ * 电荷转移激发。
在液相环境(水)中, 分别使用pbepbe/6-311++g(2d, 2p)方法和B3LYP/6-311++g(2d, 2p)方法, 优化了维生素C的AA和DHA分子结构, 并对其紫外光谱和电子激发特征进行系统的分析, 结果表明:
(1)pbepbe/6-311++g(2d, 2p)是计算维生素C的AA紫外吸收光谱更精确的方法;
(2)二面角参数表明DHA比AA的环状结构发生了显著的平面扭曲;
(3)基态跃迁到S1, S2, S3, S4, S14和S18激发态为AA产生紫外光谱的主要原因。 对AA位于200.171 5 nm处的吸收峰起主要作用的是25% S0→ S13、 15%的S0→ S14和33%的S0→ S18的跃迁, 该吸收峰包含n→ π * , n→ σ * 电子跃迁, 266.924 8 nm处的吸收峰对应38%的S0→ S1、 42%的S0→ S2、 14%的S0→ S3和5%的S0→ S4的跃迁, 该吸收峰属于n→ π * 和π → π * 的电子跃迁;
(4)基态跃迁到S6, S9, S12, S13, S15, S16, S17, S19和S20激发态为DHA产生紫外光谱的主要原因。 DHA的最强吸收峰位于181.024 8 nm处, 主要包含5%的S0→ S12、 7%的S0→ S13、 19%的S0→ S15、 23%的S0→ S16、 16%的S0→ S17、 11%的S0→ S19和11%的S0→ S20的跃迁, 该吸收峰具有n→ σ * 和n→ π * 的电子跃迁特征, 231.346 39 nm处微弱的吸收峰主要对应83%的S0→ S9的跃迁, 归属于n→ π * 电子跃迁, 282.466 8 nm处的吸收峰主要对应87%的S0→ S6跃迁, 归属于n→ π * 电子跃迁;
(5)通过空穴-电子分布及其衍生量的分析, 将AA紫外光谱起主要贡献的S4, S13, S14激发态与DHA紫外光谱起主要贡献的S6, S9, S17和S20指认为电荷转移激发, 而其他激发态的电子与空穴分离程度很低, 指认为局域激发。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|