{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函理论的肾上腺素分子的光谱分析
[陆利敏 , 施斌, 唐天宇, 赵先豪, 魏晓楠, 唐延林
, 施斌, 唐天宇, 赵先豪, 魏晓楠, 唐延林* ]
, 施斌, 唐天宇, 赵先豪, 魏晓楠, 唐延林]
|
|


作者简介: 陆利敏, 女, 1994年生, 贵州大学物理学院硕士研究生 e-mail: 2353097900@qq.com
肾上腺素是一种神经和激素的传送体, 研究肾上腺素分子的光谱和能级有助于了解其化学稳定性和药理作用。 基于密度泛函理论(DFT), 利用Gaussian 09软件在B3LYP/6-311G(d, p)基组水平上对肾上腺素分子进行结构优化, 采用含时密度泛函理论(TD-DFT)的PBE方法在def2tzvp基组水平上计算肾上腺素分子在气相中的前20个激发态, 利用Multiwfn3.7(dev)软件绘制出其紫外光谱图并对激发性质进行分析。 肾上腺素分子紫外光谱对应的主要跃迁是从基态分别到第1, 2, 4, 8, 15和16激发态的跃迁, 其他的激发态的振子强度低于阈值0.03。 理论计算得出肾上腺素的紫外光谱有两个吸收峰, 分别位于206.23和273.92 nm, 206.23 nm峰主要由基态跃迁到第16激发态形成, 273.92 nm峰主要是基态跃迁到第2、 4激发态形成, 主要是由苯环上π→π*跃迁所产生, 并与实验光谱吻合较好。 对肾上腺素分子的激发态性质分析可知, 上述吸收峰都是在最高占据轨道和最低空轨道的临近轨道跃迁产生的。 利用密度泛函的PBE方法在6-311G(d, p)的基组水平上计算肾上腺素分子频率并绘制红外光谱, 由振动分析可知, 3 738和3 662 cm-1峰是由酚羟基O—H伸缩振动产生的特征吸收峰, 3 715 cm-1峰是由醇羟基O—H伸缩振动产生的特征吸收峰, 2 854 cm-1峰是由甲基的C18—H20键的伸缩振动产生的特征吸收峰, 1 516和1 439 cm-1峰是苯环骨架的伸缩振动的特征吸收峰, 1 279与1 057 cm-1峰分别是由C6—O10和C12—O23键伸缩振动产生的特征吸收峰, 620 cm-1峰是N22—H17键摇摆振动的特征吸收峰。 对比肾上腺素的实验红外光谱, 发现理论光谱与实验光谱中各基团的特征吸收峰都较为明显且总体吻合较好。 由于肾上腺素分子二聚体和多聚体之间形成氢键, 分子间氢键的形成削弱了O—H键的强度, 降低了能形成分子间氢键的羟基O—H的伸缩振动频率, 从而导致实验光谱在3 500~2 500 cm-1之间呈现出一个宽峰。
Epinephrine is a neurotransmitter and hormone transmitter. Studying the spectra and energy levels of the epinephrine molecules may help to understand their chemical stability and pharmacological effects. Based on density functional theory (DFT), the structure of epinephrine was optimized by using Software Gaussian 09 at the level of B3LYP/6-311G(d, p) in this article. On this basis, the first 20 excited states of epinephrine molecule were calculated by using the time-dependent density functional theory (TD-DFT) at the level of PBE/def2tzvp in the gas phase, and ultraviolet spectrum was drawn by using Multiwfn3.7 (dev) software and the properties of excitation are analyzed. The main transitions corresponding to the UV spectrum of adrenaline molecules are those from the ground state to the first, second, fourth, eighth, 15th and 16th excited states respectively. The oscillator strength of other excited states is lower than the threshold value of 0.03. The theoretical calculation showed two absorption peaks in the ultraviolet spectrum of epinephrine, which are located at 206.23 and 273.92 nm respectively. The peak at 206.23 nm is mainly formed from the ground state transition to the 16th excited state, and the peak at 273.92 nm is mainly formed from the ground state transition to the 2nd and 4th excited states. They are mainly produced by the transition from π band to π* band on the benzene ring, and they are in good agreement with the experimental spectrum. The analysis of the excited state properties of adrenergic molecules shows that the absorption peaks are generated in the adjacent orbital transitions of the highest occupied orbital and the lowest vacant orbital. Then, the PBE method based on density functional theory was used to calculate the infrared frequency and draw the infrared spectrum of adrenergic molecules at the base group level of 6-311g (d, p). The vibration analysis shows that the characteristic absorption peak is generated by the phenol hydroxyl O—H vibration at 3 738 and 3 662 cm-1, and the alcohol hydroxyl O—H vibration generates the characteristic absorption peak at 3 715 cm-1. Point 2 854 cm-1 is the characteristic absorption peak generated by the stretching vibration of the C18—H20 bond of methyl, point 1 516 and 1 439 cm-1 are the characteristic absorption peaks of the stretching vibration of the benzene ring, at 1 279 and 1 057 cm-1 respectively were the characteristic absorption peak of the stretching vibration of the C6—O10 and C12—O23 bond, and point 620 cm-1 is the characteristic absorption peak of the oscillation of the N22—H17 bond. The vibration analysis is consistent with the characteristic absorption peaks of various functional groups in the introduction to spectroscopy. By comparing the experimental infrared spectrum of epinephrine, it was found that the characteristic absorption peaks of each group in the theoretical spectrum were relatively obvious, and they were generally in good agreement with the experimental spectrum. Due to the hydrogen bond formed between the adrenergic molecular dimer and the polymer, the hydrogen bond formed between the molecules weakens the strength of the O—H bond. It reduces the stretching vibration frequency of the hydroxyl O—H that can form the inter-molecular hydrogen bond, which results in a wide peak of the experimental spectrum between 3 500 and 2 500 cm-1.
肾上腺素是由肾上腺髓质分泌的主要激素[1], 作为一种重要的生物分子, 肾上腺素具有特殊的生理功能和药理性能, 其中枢作用弱而外周作用强。 在医药领域, 肾上腺素药物可以被用来作为过敏性休克的急救、 治疗心脏疾病和支气管哮喘等[2]。 作为一种生理活性物质, 肾上腺素的生理作用主要表现为对生物体的代谢作用、 各器官的作用和全身作用, 控制着神经系统进行一系列的生物反应及神经化学过程。 通过计算分析肾上腺素分子的光谱对于了解肾上腺素的生物化学性质至关重要。
由于肾上腺素是具有多种活性基团(羟基、 氨基)的功能性化合物, 对其应用和结构性能的研究一直是医学、 生物学、 化学等多门学科的热门课题。 李萍等[3]研究了复方注射液中肾上腺素及其代谢产物的高效液相色谱-电化学(HPLC-ECD)检测方法, 结果表明该方法快速简单、 选择性好、 灵敏度及准确度高, 可用于肾上腺素及其相关物质的分析测定; 张占军等[4]在 1.0 mol· L-1 H2SO4溶液中借助快速扫描循环伏安(CV)法估算出了肾上腺素在铂电极上发生电化学氧化反应时该过程的传递系数和电化学反应速率常数, 表明在快速电势扫描条件下, 肾上腺素电氧化过程遵从4G反应机理; 魏国等[5]对以氨甲环酸联合肾上腺素减少单侧全膝关节置换术后出血的有效性及安全性进行了研究, 结果显示氨甲环酸(TXA)联合肾上腺素减少单侧全膝关节置换术(TKA)后出血的疗效明显且安全; 近年来, 有报道研究了酸碱性条件下的肾上腺素表面增强拉曼(SERS)实验光谱, 但对肾上腺素分子光谱理论分析比较少。 本工作将利用Gaussian软件, 通过模拟计算来分析肾上腺素分子红外光谱、 振动类型, 紫外光谱, 以便更好地了解肾上腺素分子的结构特性, 为肾上腺素结构改造与应用提供理论依据。
使用Gaussian 09软件包, 以Gaussian View建模, 在气相环境下, 利用密度泛函(DFT)[6]的B3LYP方法在6-311G(d, p)的基组水平上对肾上腺素分子进行几何构型优化和频率计算, 结果均收敛, 无虚频且达到能量极小值点, 表明优化后结构是稳定的[7, 8, 9]。 在优化的基础上采用含时密度泛函理论(TD-DFT)[6]的PBE计算方法在def2tzvp的基组水平上计算肾上腺素分子在气相中的前20个激发态, 利用Multiwfn3.7多功能波函数软件[10, 11, 12]分析了紫外峰的跃迁模式。 最后利用密度泛函(DFT)的PBE计算方法在6-311G(d, p)的基组水平上对肾上腺素分子优化和频率计算, 用Multiwfn软件对其频率校正, 得到红外光谱, 分析肾上腺素分子红外光谱所对应的各个特征基团的振动模式。
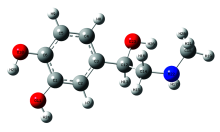
肾上腺素分子是碳, 氢, 氧, 氮四种元素构成, 其分子式为C9H13O3N, 分子量为183.204。 肾上腺素分子优化后的几何构型如图1所示, 由一个苯环、 两个酚羟基、 一个醇羟基、 一个次甲基、 一个亚甲基和一个甲氨基组成, 形成一个空间立体结构。
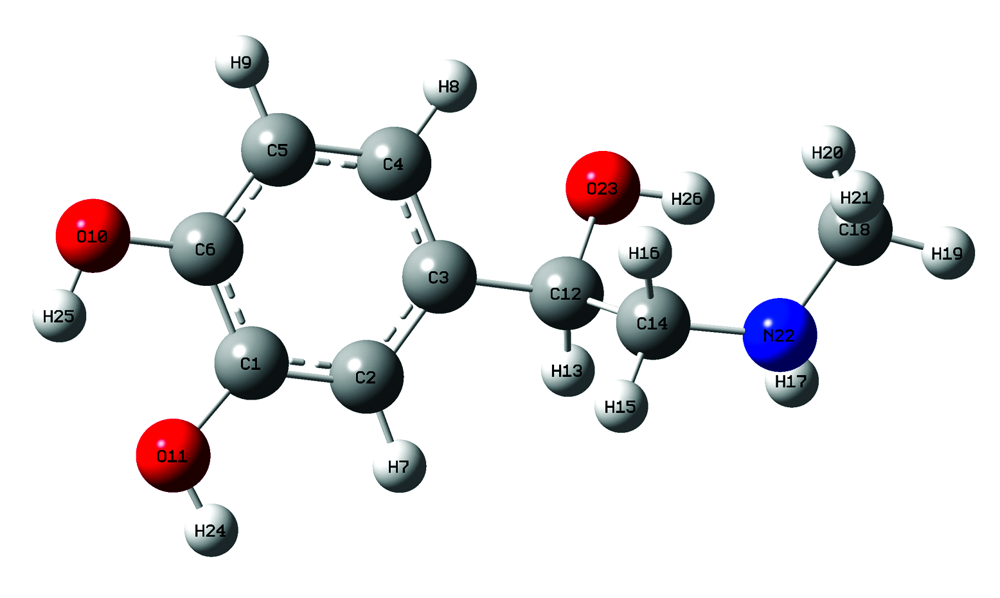 | 图1 优化后肾上腺素分子的结构Fig.1 Structure of epinephrine after optimization |
分子结构优化和频率计算的结果表明, 肾上腺素分子同时满足结构优化和频率计算的收敛标准, 优化结果收敛且频率分析无虚频, 优化结构是合理的。 优化后肾上腺素分子的能量为-630.595 295 0 Hartree。
2.2.1 紫外光谱
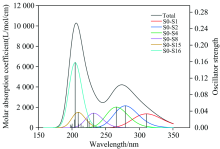
紫外光谱可以反映分子的电子结构、 能级和化学稳定性。 采用TD-DFT方法, 在优化结构的基础上计算了肾上腺素分子的前20个激发态, 并设振子强度的阈值为0.03, 得到了相应的紫外光谱如图2所示, 图中S0表示基态, Sx表示第x个激发态。 计算得出肾上腺素有两个紫外吸收峰, 波长分别为206.23和273.92 nm, 摩尔吸收系数分别为10 282.96和4 212.54 L· mol-1· cm-1, 主要由苯环中π → π * 跃迁产生。 肾上腺素分子紫外光谱对应的主要跃迁从基态分别到第1, 2, 4, 8, 15和16激发态的跃迁, 其中在206.23 nm处的峰包含了从基态到第8激发态的跃迁、 贡献率为0.81%, 基态到第15激发态的跃迁、 贡献率为14.00%, 基态到第16激发态的跃迁、 贡献率为61.31%; 273.92 nm处吸收峰包含了基态到第1激发态的跃迁、 贡献率为0.54%, 基态到第2激发态的跃迁、 贡献率为48.62%, 基态到第4激发态的跃迁、 贡献率为42.66%, 基态到第8激发态的跃迁、 贡献率为0.06%。 其他强度低于阈值0.03的跃迁就不一一分析。 理论计算得出的紫外光谱图与中国科学院上海有机化学研究的化学专业数据库[13]的实验谱图对比吻合较好。
 | 图2 肾上腺素分子紫外光谱Fig.2 UV spectrum of epinephrine molecule |
紫外吸收光谱是由于价电子的跃迁而产生的。 紫外光谱吸收带的位置与所吸收的光的波长相对应, 即与电子跃迁吸收的能量相对应, 而吸收带的强度在很大程度取决于吸收光的能量与分子由基态跃迁到激发态这一电子体系之间互相作用的几率和激发态的极性。 表1给出了肾上腺素分子从基态跃迁到各个激发态的所对应的能量间隔、 紫外光谱波长、 振子强度、 轨道跃迁及各轨道跃迁的权重。 肾上腺素分子的最高占据轨道(HOMO)和最低未占据轨道(LUMO)分别为第49电子轨道和第50电子轨道, 从表1中还可以看出这些跃迁都是在最高占据轨道和最低未占据轨道附近轨道间的跃迁。 在较强的6个激发态中, 从基态到第16激发态跃迁的振子强度最大。
| 表1 肾上腺素分子激发态的性质 Table 1 Properties of excited states of epinephrine molecule |
2.2.2 红外光谱
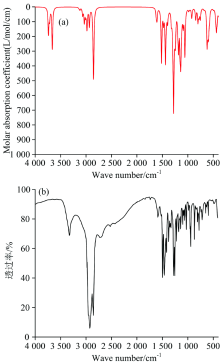
分子红外光谱吸收峰的频率、 强度及形状都与分子的结构密切相关。 红外光谱峰的位置由振动频率决定, 化学键的振动频率与键的力常数和成键原子的折合质量有关[14], 键的力常数越大, 表明该键越稳定, 对应的振动频率越高。 利用密度泛函(DFT)的PBE方法在6-311G(d, p)的基组水平上对肾上腺素分子优化和频率计算, 计算得到无虚频的情况下, 再利用Multiwfn3.7软件对所有频率进行校正, 校正因子为0.99, 并绘制气相下肾上腺素分子4 000~400 cm-1 的红外光谱图并与实验谱图进行比较如图3(a, b)示, 其特征吸收峰的振动归属分析如表2。 在3 738和3 662 cm-1吸收峰是由酚羟基O—H伸缩振动产生的特征吸收峰, 3 715 cm-1峰是由醇羟基O—H伸缩振动产生的特征吸收峰, 2 854 cm-1峰是甲基的C18—H20键伸缩振动产生的特征吸收峰, 1 516和1 439 cm-1处峰是由苯环骨架的伸缩振动产生的特征吸收峰, 1 279与1 057 cm-1处峰分别是由C6—O10和C12—O23键伸缩振动产生的特征吸收峰, 620 cm-1处峰是N22—H17键摇摆振动产生的特征吸收峰。
 | 图3 肾上腺素分子的红外光谱 (a): 理论图; (b): 实验图Fig.3 Infrared spectra of epinephrine molecule (a): The theory of figure; (b): The experimental figure |
| 表2 肾上腺素分子红外峰对应的主要振动归属 Table 2 Major vibration attribution corresponding to infrared peaks of epinephrine molecule |
对比肾上腺素分子红外光谱的理论计算图与实验图(中国科学院上海有机化学研究所化学专业数据库), 可知两者特征吸收峰基本吻合, 但在3 000 cm-1以上特征吸收峰有较大的差异。 理论图在3 650~3 750 cm-1有O—H的特征吸收峰, 而实验谱图在3 500~2 500 cm-1处出现了一个宽峰, 这是肾上腺素分子二聚体和多聚体之间形成了不同强弱的氢键, 形成的分子间氢键削弱了原分子中酚羟基O—H键和醇羟基O—H键的强度, 导致其伸缩振动频率减小, 从而导致实验光谱在3 500~2 500 cm-1之间呈现出一个宽峰。
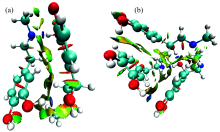
为了探究肾上腺素分子键能否形成氢键, 用genmer软件和molclus1.9.6 软件[15]搜索了20个肾上腺素二聚体构象和5个肾上腺素三聚体构象, 对其进行结构优化, 优化无虚频之后再通过Multiwfn3.7软件和VMD软件绘制出肾上腺素二聚体和三聚体的RDG函数等值面图, 分别如图4(a, b)所示, 图中蓝色等值面表示的是氢键、 绿色表示范德华作用和棕色表示的是弱位阻作用, 发现有16个二聚体构象和5个肾上腺素三聚体构象中都存在不同强度的氢键, 且肾上腺素二聚体和多聚体中是—NH和—OH基团形成了氢键, 从而导致—OH基团的红外吸收频率会向低频方向移动。
 | 图4 肾上腺素二聚体与三聚体RDG函数等值面图 (a): 二聚体构象; (b): 三聚体构象Fig.4 RDG function isosurface map of dimer and trimer (a): Dimer conformation; (b): Trimer conformation |
肾上腺素是一种神经和激素的传送体, 研究肾上腺素分子的光谱和能级有助于了解其化学稳定性和药理作用。 同时也为肾上腺素分子的实验光谱奠定理论基础。 本理论研究表明: 肾上腺素分子紫外光谱对应的主要跃迁是从基态分别到第1, 2, 4, 8, 15和16激发态的跃迁, 其他的激发态的振子强度低于阈值0.03。 理论计算得出肾上腺素的紫外光谱有两个吸收峰, 波长分别位于206.23和273.92 nm, 206.23 nm峰主要由基态跃迁到第16激发态形成, 273.92 nm的峰主要由基态跃迁到第2、 4激发态形成, 它们主要由苯环上π → π * 跃迁所产生, 并与实验光谱吻合较好。 对肾上腺素分子的激发态性质分析可知, 上述吸收峰都是在最高占据轨道和最低空轨道的临近轨道跃迁产生的。 在红外谱图中, 3 738和3 662 cm-1处的吸收峰是由酚羟基O—H伸缩振动产生的特征吸收峰, 3 715 cm-1处峰是由醇羟基O—H伸缩振动产生的特征吸收峰, 2 854 cm-1处峰是甲基的C18—H20键伸缩振动产生的特征吸收峰, 1 516和1 439 cm-1处峰是由苯环骨架伸缩振动产生的特征吸收峰, 1 279与1 057 cm-1处峰分别是由C6—O10和C12—O23键伸缩振动产生的特征吸收峰, 620 cm-1处峰是N22—H17键摇摆振动产生的特征吸收峰。 对比肾上腺素的实验红外光谱, 发现理论计算光谱与实验光谱中各基团的特征吸收峰都较为明显且总体吻合较好。 肾上腺素分子二聚体和多聚体之间形成的分子间氢键削弱了单个肾上腺素分子中O—H键的强度, 降低了其伸缩振动频率, 从而导致实验光谱在3 500~2 500 cm-1之间呈现出一个宽峰。 本研究对更深入了解肾上腺素的生化活性、 化学稳定性和神经递质作用有一定的帮助。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|