
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
高煤级煤石墨化轨迹阶段性的XRD和Raman光谱表征
[李焕同1, 2  , 曹代勇
, 曹代勇3, * , 张卫国1, 2 , 王路4 ]
, 曹代勇, 张卫国|
|


作者简介: 李焕同, 1986年生, 西安科技大学地质与环境学院讲师 e-mail: htlcumt@126.com
为了研究石墨化过程中煤的分子结构有序化轨迹, 选取湖南、 陕西19个不同变形-变质程度高煤级煤为研究对象, 采用工业分析、 元素分析、 X射线衍射分析(XRD)和拉曼光谱分析(Raman)等手段, 结合分峰拟合的数学方法, 对系列样品分子结构参数(XRD结构参数, 如石墨化度、 延展度 La、 堆砌度 Lc及面网间距 d002等; Raman参数, 如 PG, P1, R1, R2等)进行了统计与计算。 研究结果表明: 煤化作用阶段H/C随变质程度增加而逐渐减小, 但在石墨化阶段以物理变化为主, 其趋势变缓或不显著; XRD参数 d002, La, Lc, N及 La/ Lc等随变质程度呈现非线性连续(阶跃性)变化, 拐点大致对应 Rm=7.0%, d002=0.338 nm, 拐点之前 La, Lc及 N变化较小(或平稳增大), 拐点之后石墨晶体结构快速形成, 微晶尺寸增大, 拼叠作用开始并逐渐增强; La/ Lc变化亦反映石墨化过程由缩合作用向拼叠作用转变。 高煤级煤石墨化轨迹可按有序化增加的三阶段模型来表述, 无定形碳(无烟煤)至变无烟煤阶段, G峰位、 峰位差 P1变化显著, ID1/ IG在表达无序程度时不服从TK关系; 变无烟煤至半石墨阶段, 即从石墨化开始结构演化轨迹呈现不同方向, R1随着有序的增加呈现截然相反的轨迹, 部分石墨组分演化服从TK关系, R2在石墨化度为45%时呈现截然相反的轨迹; 石墨阶段温、 压作用导致微晶尺寸急剧增加(或阶跃), ID1/ IG减小服从TK关系。 当不同石墨化程度的新生组分共存时, d002不足以代表样品最大的演化程度, 但其作为平均度量来标度高煤级煤石墨化过程中结构演化特征仍为较优的选择, 且(002)和( γ)峰半峰宽能较好地区分石墨化煤的变质类型, H/C, ID1/ IG亦随 d002演化轨迹不同, 需利用 d002<0.344 0 nm, R1<2.0, H/C<0.12等综合指标判别石墨化的开始。 由此可以看出, 采用XRD和Raman光谱分析技术可以表征高煤级煤石墨化轨迹阶段性以及结构的差异性。
In order to the interpretation of ordering and crystallinity of natural graphitized coal, nineteen kinds of different deformation-metamorphism degree high-rank coal from Hunan Province and Shaanxi Province were studied with proximate and ultimate analysis, X-ray diffraction (XRD), Raman spectrum and curve-fitting analysis. The graphitization, crystal size ( La and Lc), interplanar spacing ( d002) were calculated with XRD. The parameters of PG (G band position), P1 (G and D1 band separation), R1= ID1/ IG, the peak height ratio, R2= AD1/( AG+ AD1), peak area ratio were calculated with Raman. The results showed that the H/C decreases gradually with the increase of metamorphic degree during the coalification stage, but during the graphitization stage, the change was primarily physical, and the trend was slow or not significant. The parameters of d002, La, Lc, N and La/ Lc had shown that the crystalline structure of natural graphitized coal presented nonlinear continuous (step) change with metamorphism degree. The inflection point corresponds roughly to Rm=7.0% and d002=0.338 nm. Before the inflection point, La, Lc and N changed little (or increase steadily), and the graphite crystal structure formed rapidly after the inflection point, the stacking effect begins and gradually increases, as the crystallite size increases. La/ Lc variation reflected that the graphitization process changed from condensation to overlap. The graphitization trajectory of high-rank coal can be given in a three-stage model of orderly increase. During the stage from amorphous carbon (anthracite) to meta-anthracite, the parameters of PG and P1 changed significantly, and ID1/ IG did not obey the TK relation when expressing the degree of disorder. During the stage from meta-anthracite to semi-graphitization showed different directions, R1 presented an opposite trajectory with the increase of order, the evolution of some graphite components followed the TK relation, and R2 showed a completely contradictory trajectory when the graphitization degree was 45%. The temperature and pressure in the graphite stage led to a sharp increase in crystal size (step evolution), and the decrease of ID1/ IG obeying the TK relation. As neogenesis-associated components in different graphitized coals, d002 cannot reflect the largest metamorphic degree of graphitized coal. However, it was still a superior choice to consider d002 as an average scaling of the graphitized coals in the process of graphitization. Moreover, full width at half maximum of the (002) and ( γ) band are reliable indicators for distinguishing and classifying of metamorphism type of nature graphitized coals. H/C, and ID1/ IG also evolved over d002 trajectory was altered, needed to use d002<0.344 nm, R1<2.0, H/C<0.12 and other comprehensive indicators to identify the beginning of graphitization. From this, it could be seen that XRD and Raman spectral analysis techniques could be used to characterize the graphitization track stages and structural differences of high rank coal.
煤中有机质演化是连续性过程, 石墨化作用作为煤化作用的延续, 其趋势是结构有序化、 化学成分单一化(增碳脱氢及异种元素排出的过程), 结构缺陷消亡, 最终演变为以碳元素为主、 三维有序结构的集合体(煤系石墨)[1, 2, 3]。 当煤的镜质体最大反射率Rm> 8%时, 基本结构单元延展度剧增, 标志着煤化作用的结束及石墨化作用的开始[4, 5]。 尤其在岩浆热变质及构造应力作用下, 物理、 化学和结构表现出的连续和渐进的石墨化过程[2, 6], 或具有中间结构状态的不连续过程(跃变)[2, 7]。 通过XRD估算的石墨化程度是变质级别的可靠近似指标[8], 并且碳物质在足够高的温度和压力下都将转化为石墨, 由面网间距d002=0.344 nm开始, 乱层结构向有序结构演化[9], 也被视为石墨化的起点。 Raman光谱是研究无序碳材料的有力方法, 具有需要少量样品和原位分析的优点, 从而减少结构信息的损失, 同样其也适用于天然石墨化煤的石墨化程度的定量表征[10, 11], 以揭示芳香碳环中原子与分子的振动信息, 反映结构有序程度和结构缺陷[12]。
高煤级煤中芳香晶核经历芳构化、 环聚合、 拼叠作用和秩理化作用[1], 向石墨结构发展的过程, 非定向的芳香碳经过一系列的微观结构和化学成分的变化产生各种中间相态, 有序轨迹演化可能存在阶段性区间。 本文以湖南、 陕西高煤级煤为研究对象, 借助元素分析、 XRD及Raman等测试结果, 探讨不同演化段的有机质结构参数的特征及演化轨迹, 以期对高煤级煤分级分质利用、 煤炭由燃料向工业原料转变提供借鉴。
煤样采自湖南寒坡坳矿区(H1— H11), 五峰仙矿区(W12— W15), 以及陕西凤县岩湾矿区(Y16— Y19), 共19个不同煤级、 不同变形程度的岩浆热变质煤样(表1), 均为井下全层样。 采样方法遵照GB/T 482— 2008《煤层煤样采取方法》执行, 样品采集后及时装入塑料样袋中封存, 以避免污染。
| 表1 各煤样的工业分析、 元素分析与镜质组最大反射率 Table 1 Vitrinite reflectance, proximate and ultimate analysis of samples |
手工选取新鲜煤样, 其中粉碎粒度小于0.1 mm的煤样, 用于镜质体反射率测试, 样品制备符合GB/T 16773— 2008《煤岩分析样品制备方法》规定。 另取200目(74 μ m)的煤样10 g放入聚四氟乙烯烧杯中, 加入30 mL浓盐酸, 在搅拌状态下浸泡24 h, 在电热板上煮沸蒸至近干, 加入蒸馏水稀释冲洗, 沉淀后去掉上部清液, 反复数次至中性, 滤出煤样; 在经盐酸处理后的样品中加入浓盐酸和浓氢氟酸的混合溶液50 mL, 重复上述步骤; 最后将滤出的煤浆放入干燥箱中24 h(温度65 ℃)烘干, 研磨后用于XRD及Raman测试。
煤中水分、 灰分和挥发分依据GB/T 212— 2008《煤的工业分析方法》测定; 煤中元素依据GB/T 31391— 2015《煤的元素分析》测定; 煤镜质体反射率依据GB/T 6948— 2008《煤的镜质体反射率显微镜测定方法》测定。
采用MSAL-XD2 X射线衍射仪上进行, Cu靶, K辐射, 管压36 kV, 管流30 mA, 发散狭缝1 mm, 接收狭缝0.16 mm, 步进式扫描, 步宽0.02° , 扫描速度2° · min-1, 测量范围为5° ~70° 2θ , 进行样品大分子结构测定。 采用LabRam HR Evolution型光谱仪对样品进行Raman测试, 激光波长514.5 nm, 扫描范围400~4 000 cm-1, 由于煤样的非均质性, 每次测试都在样品的3~10个不同位置进行, 再将所测结果取平均值。
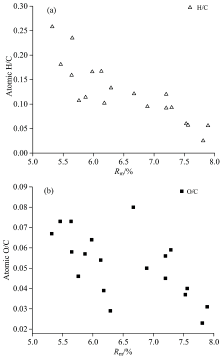
新生石墨化组分随着石墨化程度增高而增多, 影响了反射率测定的规律性和灵敏度, 各煤样镜质组最大反射率(Rm)变化于5.46%~7.89%, 反射值较低, 均值为6.55%。 灰分变化于1.23%~13.30%, 均值为7.96%(除SXL100); 挥发分变化于4.21%~6.95%, 均值为5.13%(表1); 固定碳变化于51.92%~93.50%, 均值80.92%; 煤化作用阶段以降解和芳构化为主, 氢、 碳原子数目比(H/C)随着Rm的增加降低, 进入石墨化阶段这一过程转变为碳结构层的有序化, H/C降低趋势变缓或不显著[图1(a)], 而氧、 碳原子数目比(O/C)在Rm=6.5%~7.0%前后呈阶段性演化[图1(b)]。
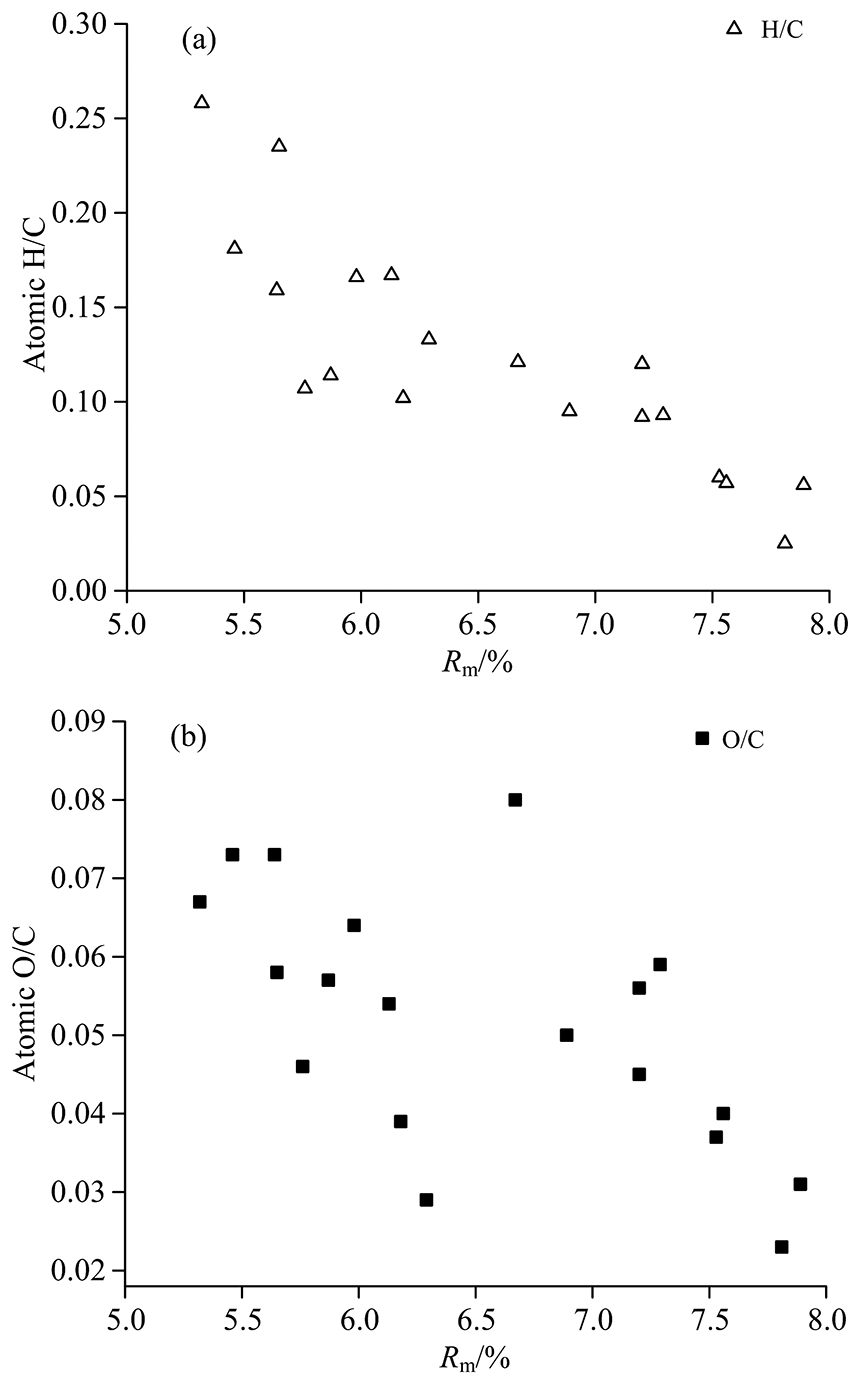 | 图1 各煤样的H/C(a), O/C(b)原子数目比与Rm关系图Fig.1 Correlation of H/C (a), O/C (b) atomic ratios and vitrinite reflectivity (Rm) |
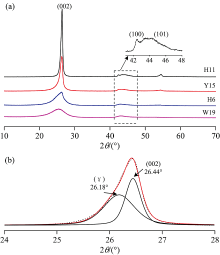
XRD可以揭示“ 微晶” 结构排列的规则性。 通常XRD谱图中~26° 2θ 处的宽缓峰被认为是(002)峰和γ 带叠加产生(图2), (002)峰反映芳香结构单元的平行定向程度, γ 带与分子中脂肪碳(脂链和脂环)结构相关, 变质程度越低, 脂肪碳结构越发育[13], 相反, 随着变质程度升高, (002)峰逐渐收窄, 强度变强, 反映芳香层结构逐渐规整、 有序; 44° 2θ 处为(100)峰和(101)峰的叠合峰。 通过分峰拟合可得到相应的峰位、 衍射强度及半峰宽等参数(表2), 利用Bragg方程和Scherre公式[14], 计算出基本结构单元结构参数芳香层面网间距d002、 堆砌度Lc和延展度La。
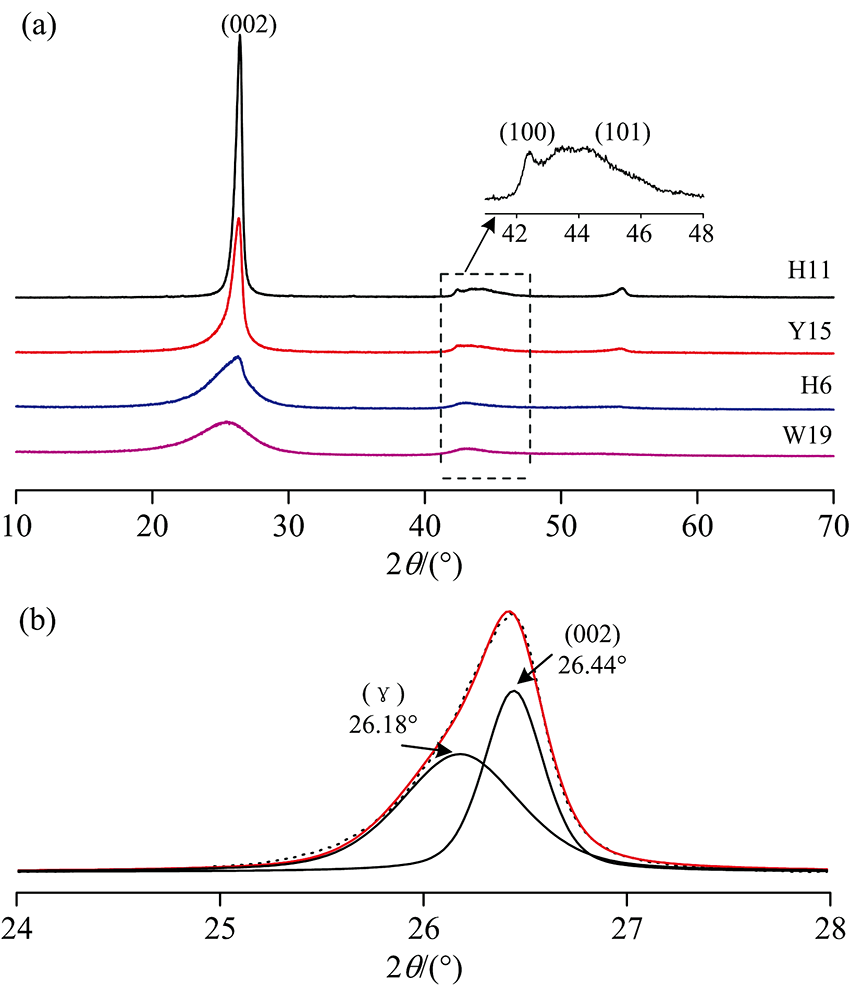 | 图2 (a)典型样品的XRD谱图; (b)(002)衍射峰分峰拟合, (γ )峰为无定形碳, (002)峰为石墨化碳Fig.2 (a) X-ray diffraction profiles of the representative de-mineralized samples; (b) Curve-fitting of two Lorentz-Gaussian peaks for H11 coal at 26.44° (002-band) and 26.18° (γ -band) in 2θ range 24° ~28° , signifying the number of crystalline carbon and aromatic carbon, respectively. The dots and the red line indicate the observed intensities and generated intensities respectively |
| 表2 各煤样XRD谱图(γ )、 (002)峰峰位和半峰宽以及结构参数 Table 2 Peak positions, full width at half maximum (FWHM), and obtained structural parameters extracted from curve-fitting of (002), (γ ) bands for samples of XRD spectra |
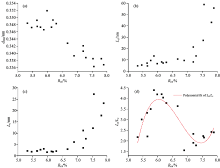
图3显示, d002随Rm增高而减小, La, Lc与堆砌层数N随Rm增高而增大[图3(a, b, c)], 说明随着变质程度的增加, 煤中微晶尺寸逐步增大。 然而, 当Rm< 7.0%时d002, Lc和La变化较小, 之后呈现缓慢增大(平稳)至急剧增大的突变(或跃变)[图3(b, c)]。 La与煤受热温度有关, 较高的温度有利于La的增大, 而较高的压力有利于Lc增大[1], 静压力和挤压力均有利于芳香层片的叠加[2]。 图3(d)显示, 高煤级煤石墨化结构演化轨迹的变化, 在热力条件下, La和Lc稳定增加, N由5~6层, 增大到6~10层, 但La/Lc值迅速增加, 此阶段结构演化主要形式仍是环缩合作用; 在较高温度和定向压力作用下, La和Lc迅速增加, N达到10层以上, La/Lc值在较低的范围内增加, 此阶段结构演化显示拼叠作用开始并逐渐增强。
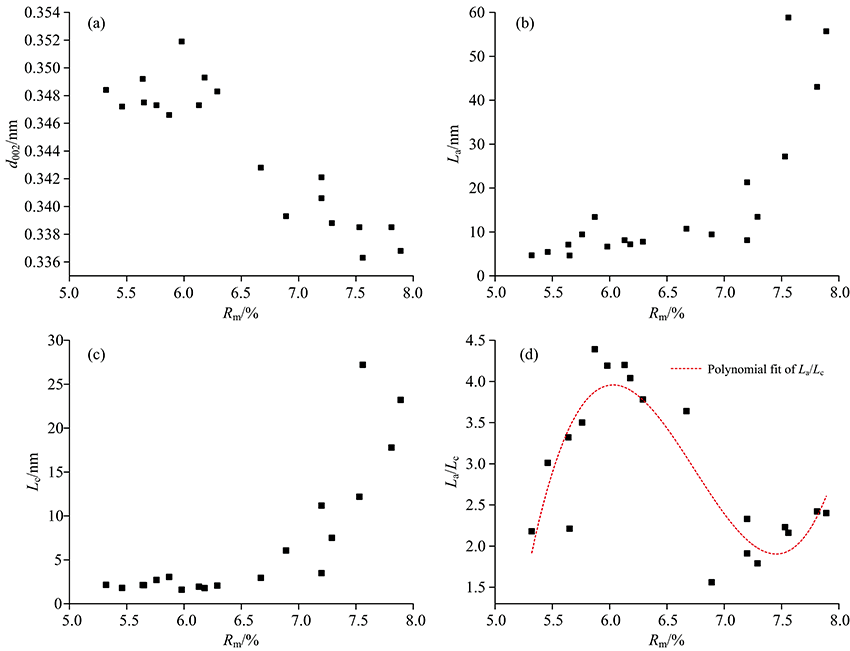 | 图3 石墨化煤的XRD参数与镜质组反射率(Rmax)相关分析图Fig.3 Correlation between XRD parameters and vitrinite reflectivity (Rmax) for serial graphitized coals |
常用结构参数d002划分石墨(< 0.338 nm)、 半石墨(0.338~0.340 nm)、 变质无烟煤(0.340~0.348 nm)和无烟煤(> 0.344 nm), 并且常用的“ 石墨化度” (即无定形碳演化接近完美石墨的程度)实质上也是依据d002计算的, 其公式为: G.D (graphitization degree)=(0.344 0-d002)/(0.344 0-0.335 4) (Mering and Marie, 1958)。 然而, 即使H9与Y19石墨化程度一致(表2), 其微晶尺寸(La, Lc及N)却有较大不同, 所以平均面网间距d002不足以代表样品最大的演化程度, 尤其是不同石墨化程度的新生组分共存时。 但是, 煤系石墨的形成是自身物质组成、 温度、 压力和矿物催化等因素综合效应[2], 其作为平均度量来标度高煤级煤石墨化过程中结构演化特征仍不失为较优的选择(图6)。
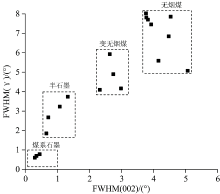
随石墨化煤变质程度升高, 由于新生石墨组分增多, 测得的反射率值偏低, 利用其无法有效地划分石墨化煤的变质类型。 如前文对比H9与Y19样品的d002与石墨化程度相同, 但它们的微晶尺寸和堆砌层数有较明显的差异, Y19平均堆砌层数大于50层, 说明Y19石墨微晶结构接近三维有序。 因此, 利用(002)峰半峰宽反映的石墨结晶程度与(γ )峰反映的结构无序程度, 能较好地区分系列石墨化程度煤(图4), Y19样品归为煤系石墨, 而H9中高度无序化的无定形碳含量较多, (γ )峰半峰宽较大为1.85° 2θ 。 XRD参数反映样品整体的平均结构, 参数对比演化趋势显示非线性连续(阶跃)变化, 但煤系石墨结构的不均一性、 缺陷或有序化差异, 即使Raman光谱表征平均结构的演化时也可能存在其他途径。
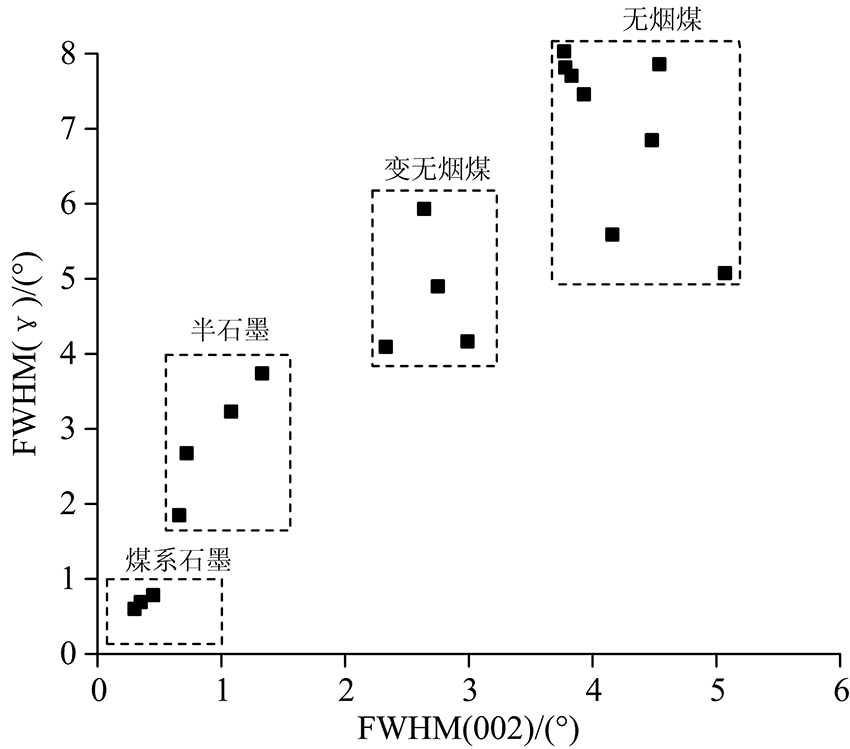 | 图4 利用(002), (γ )峰半峰宽对石墨化煤的划分Fig.4 Grouping of graphitized coals according to (002) and (γ ) FWHM |
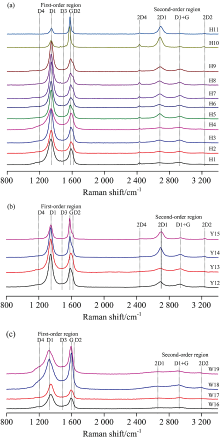
拉曼光谱是碳材料的标准表征技术。 一级模拉曼光谱包括D1峰(1 325~1 348 cm-1), D2峰(1 609~1615 cm-1), D3峰(1 483~1 554 cm-1), D4峰(1 137~1 221 cm-1)和G峰(1 570~1 596 cm-1), 完好石墨结构的拉曼光谱一级模只显示位于1 580 cm-1附近与E2g振动模相应的G峰(亦称石墨峰), 属于芳香族面域内环与链中所有sp2晶格格位的伸缩振动,
 | 图5 各煤样Raman光谱特征Fig.5 Raman spectra are composed of first-order regions, with the main peaks suggested (graphite G peak and D1 to D4 defect-activated peaks), and corresponding second-order regions resulting from overtone scattering |
| 表3 各煤样Raman光谱参数 Table 3 Raman spectroscopic parameters for the graphitized coals |
Tuinstra和Koenig (1970) 开始应用拉曼光谱研究石墨碳, 并首次获取了石墨拉曼光谱并发现微晶尺寸(芳香层片延展度La, 由XRD估算), 缺陷峰(D1峰)与石墨G峰的强度比之间呈反比(TK公式), 即ID1/IG∝ 1/
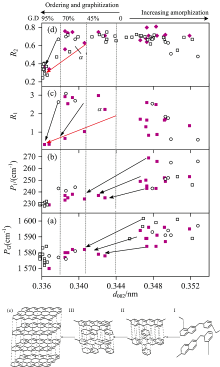
 | 图6 拉曼光谱参数随面网间距d002变化的阶段模型 ■: 本文; □: 数据源于[3]; ○: 数据源于[10]Fig.6 Raman spectroscopy parameters versus d002 (a): PG (G band position); (b): P1 (G and D1 band separation); (c): R1=ID1/IG, peak height ratio; (d): R2=AD1/(AG+AD1), peak area ratio; (e): structure model |
如图6所示, 自然条件下高煤级煤石墨化过程中分子结构有序化增加可按三阶段模型来描述[图6(e)], 先从无定形碳到石墨化无烟煤, 再到半石墨, 最终到有序石墨晶体。 概括地说, 阶段I相应于温度诱导下芳香层片延展度La逐渐增大, 成键主要为sp2, 可见光拉曼光谱中可完全忽略C— H模式。 平均G峰位置从1 600 cm-1大致变到1 585 cm-1[图6(a)], 平均峰位差P1从270 cm-1大致变到240 cm-1[图6(b)]。 D峰的出现和其强度增加服从R1(ID1/IG)∝
阶段Ⅱ 由乱层结构向有序结构演化, 相应于面网间距(d002)由0.344 nm开始石墨化[9], 芳香层片延展度Lc增速较大, 即相对温度作用下的应力诱导增强, 参与三维晶体堆砌的芳香层片层数增多。 此阶段平均G峰位置变化于1 585~1 575 cm-1, 平均峰位差在235 cm-1附近变动。 实际上, 非定向的芳香碳经历一系列的物理、 化学结构演变产生各种中间相态, 残留煤岩显微组分和新生的石墨组分及热解炭共存。 随着变质程度(温压条件)升高, 石墨组分含量逐渐增多, 并且温压作用导致石墨化速率、 产物不同, 表现为无序结构与有序石墨微晶混成不均一的集合体, 致使结构演化轨迹呈现不同方向, 即从石墨化开始, R1(ID1/IG)随着有序的增加呈现截然相反的轨迹[图6(c)], 部分样品(红色箭头)演化服从TK关系; R2(AD1/(AG+AD1))在石墨化度为45%时(d002=0.340~0.341 nm)呈现截然相反的轨迹[图6(d)]。 显然, 这种变化与样品中石墨化组分数量直接相关, 夹角α 和α '大小一定程度上可代表温度、 压力等作用强度导致有序轨迹偏离程度。
阶段Ⅲ 演变为最终在热动力条件下具有更稳定的ABAB型有序排列的三维石墨结构, 此阶段温、 压作用导致堆叠度增加和结晶尺寸急剧增加[图3(b, c)], 通常理解为石墨晶体结构快速形成(突变或阶跃)。 平均G峰位置< 1 585 cm-1, 平均峰位差< 2 35 cm-1。 D峰出现和其强度的减小服从TK关系。 d002与H/C原子数目比有较好的正相关关系[图7(b)], 亦存在高度石墨化煤H/C低, 但有序化程度并不高; 图7(a)显示H/C随ID1/IG演化的不同轨迹, 在R1< 2.0, H/C< 0.12时开始石墨化, 即使在之后阶段H含量减小, 并不意味着其芳香层片的定向性很好。
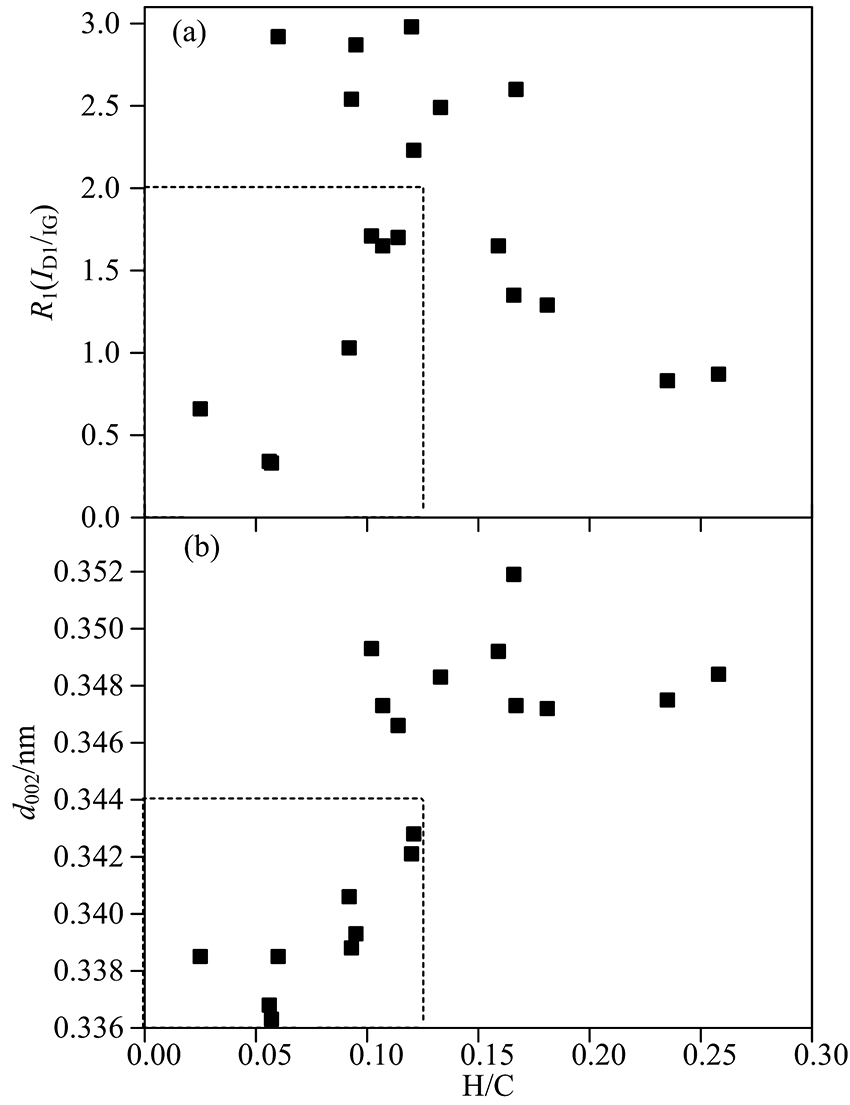 | 图7 ID1/IG, d002与H/C原子数目比的关系Fig.7 Correlation of ID1/IG, d002 and H/C atomic ratio for serial graphitized coals |
(1)高煤级煤石墨化过程是化学成分单一化、 分子结构有序化的过程, H/C随变质程度增加而逐渐减小, 但在石墨阶段趋势变缓或不显著。
(2)XRD参数表征微晶结构平均尺寸大小, d002, La, Lc, N及La/Lc等随变质程度呈现非线性连续(阶跃性)变化, 拐点大致对应Rm=7.0%, d002=0.338 nm, 拐点之前La, Lc及N变化较小(或平稳增大), 拐点之后石墨晶体结构快速形成; La/Lc变化反映石墨化过程由缩合作用向拼叠作用转变。
(3)高煤级煤石墨化可按有序化增加的三阶段模型来表述, 无定形碳(无烟煤)至变无烟煤阶段, G峰位、 峰位差P1变化显著, ID1/IG在表达无序程度时不服从TK关系。 变无烟煤至半石墨阶段, 即从石墨化开始结构演化轨迹呈现不同方向, R1随着有序的增加呈现截然相反的轨迹, 部分石墨组分演化服从TK关系; R2在石墨化度为45%时呈现截然相反的轨迹。 石墨阶段温、 压作用导致微晶尺寸急剧增加(突变或阶跃), ID1/IG减小服从TK关系。
(4)不同石墨化程度的新生组分共存时, d002不足以代表样品最大的演化程度, 但其作为平均度量来标度高煤级煤石墨化过程中结构演化特征仍为较优的选择, 且利用(002)和(γ )峰半峰宽能较好地区分石墨化煤的变质类型, H/C随d002, ID1/IG演化轨迹不同, 利用d002< 0.344 0, R1< 2.0, H/C< 0.12等综合指标判别石墨化的开始。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|