

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
啶虫脒太赫兹光谱的实验研究和理论模拟
[张同军 , 李德华, 曹秋红, 林红梅, 郝建军]
, 李德华, 曹秋红, 林红梅, 郝建军]
, 李德华, 曹秋红, 林红梅, 郝建军]
|
|


作者简介: 张同军, 1976年生, 山东科技大学电子信息工程学院博士学位讲师 e-mail: supoptimal@163.com
啶虫脒是一种氯吡啶类新烟碱类杀虫剂, 由于其对昆虫烟碱乙酰胆碱受体的结合选择性而成为最常用的杀虫剂之一。 为研究啶虫脒在太赫兹波段的指纹特征与其结构信息之间的关系, 利用太赫兹时域光谱(THz-TDS)技术并结合密度泛函理论(DFT)对啶虫脒晶体的太赫兹吸收光谱进行了实验研究和理论模拟。 利用THz-TDS技术测量了室温下啶虫脒在0.3~3.3 THz频段的特征吸收谱, 发现啶虫脒晶体在该频段内有多处强度不同的特征吸收峰, 分别位于1.08, 1.38, 1.97, 2.54, 2.89 THz处, 这些特征吸收峰构成了用于检测啶虫脒的THz指纹谱。 为更好地理解啶虫脒太赫兹实验特征谱产生的理论机理, 基于密度泛函理论分别对啶虫脒孤立分子模型和晶胞模型进行了理论模拟计算。 单分子模型的理论计算在Gaussian09软件中进行, 采用基于密度泛函理论的B3LYP杂化泛函方法, 并选取6-311G(d, p)基组进行几何优化, 并在相同水平上进行振动频率计算, 模拟结果与实验数据存在一定的差异, 说明单分子模拟存在一定的局限性。 晶胞模型的理论计算在Materials Studio 8.0中适合计算周期性结构的CASTEP模块中进行, 采用基于平面波赝势和广义梯度近似(GGA)的PW91, PBE, PBEsol和WC四种交换相关泛函对啶虫脒晶胞模型进行几何优化和晶格动力学计算。 将啶虫脒单分子和晶胞的理论模拟结构参数(键长、 键角)分别与其X射线衍射(XRD)实验测量的结构参数进行了详细的比较分析, 发现基于PBE方法获得的固态仿真结果中分子的结构参数与X射线衍射实验数据的一致性最好。 同时用PBE方法获得的理论仿真谱与实验吸收谱也最为吻合。 基于PBE的计算结果对实验特征吸收峰进行了振动模式指认。 研究结果表明, 啶虫脒晶体在太赫兹频段的特征吸收主要来源于晶体中以C—H…N氢键为主的分子间弱相互作用带动的集体振动模式, 以及由分子内吡啶环的平动和甲基基团的扭动所引发的骨架振动。
Acetamiprid is a chloropyridine neonicotinoid insecticide that is one of the most commonly used pesticides due to its binding selectivity toward the nicotinic acetylcholine receptor of insects. To understand the relationship between the fingerprint feature and the corresponding structural information in the terahertz region, experimental and theoretical investigations of the terahertz absorption spectrum of acetamiprid crystal were carried out using the terahertz time-domain spectroscopy (THz-TDS) and density functional theory(DFT). The terahertz absorption spectrum of acetamiprid was measured in 0.3~3.3 THz frequency range at room temperature by the THz-TDS system. A number of characteristic absorption peaks in this range were observed at 1.08, 1.38, 1.97, 2.54 and 2.89 THz, respectively, which can be the THz fingerprint spectrum for detecting acetamiprid. To better understand the experimental absorption spectrum's theoretical mechanism, calculations based on density functional theory were performed to analyze the isolated molecule and unit cell of acetamiprid. The geometry optimization and frequency calculation of the isolated molecule model was performed using DFT with periodic boundary conditions employing the B3LYP hybrid functional with the 6-311G(d,p) basis set. Some differences were observed between the simulation results and the experimental data, which means that the isolated molecule simulation has some limitations. The theoretical calculations of the unit cell were performed based on the solid-state DFT using the CASTEP program, a part of Materials Studio 8.0 from Accelrys. The calculations were performed on the crystalline state within the generalized gradient approximation (GGA) at PW91, PBE, PBEsol and WC correlation functions. The calculated structural data of bond lengths and bond angles for acetamiprid molecule and unit cell were compared with -X-ray diffraction values(XRD). Among these calculations, PBE simulations provide a significantly similar tendency with the XRD experimental values. And the calculated THz spectrum of the PBE provides better agreements with observed THz spectral characters. Consequently, the observed spectral features were assigned according to the results of the PBE simulation. The study indicates that acetamiprid's characterized features primarily originated from intermolecular collective vibrational modes, which were dominated by hydrogen bonds such as C—H…N.
太赫兹波是电磁波谱上位于微波与红外之间, 频率在0.1~10 THz, 波长在0.03~3 mm范围内的电磁辐射。 相比传统的光谱检测技术, 太赫兹时域光谱(Terahertz time-domain spectroscopy, THz-TDS)技术在特征吸收、 光子能量、 灵敏度以及相干性等方面具有独特的优势。 近年来, 太赫兹技术在公共安全、 化工检测、 物质鉴别、 生物医学等诸多领域应用广泛[1], 展现出重大的科学价值和诱人的应用前景。 THz-TDS不仅可以实现对物品的快速无损检测, 而且能够获得物质在分子水平上的结构信息和功能特性[2]。 目前THz-TDS已经成为一种可以应用于有机分子检测与研究的新型光谱技术[3]。
有机农药晶体中分子间的氢键、 范德华力等弱相互作用, 晶格的声子振动、 骨架振动以及分子内的低频集体振动模式等正好出现在THz频段内[3], 使得利用太赫兹技术研究农药的特征吸收、 分子结构、 晶体结构成为可能, 吸引了众多国内外研究者的兴趣。 Wang等[4]借助量子化学软件对绿麦隆除草剂的THz谱进行了研究, 准确解析了该农药的简正振动模式; 有研究对三种菊酯农药进行了定性和定量检测, 含量检出限达2.0%; 有报道使用模式识别算法对三种PGR农药进行了分类检测, 结果显示具有较好的鲁棒性; Qin[5]结合超材料对多菌灵农药的THz谱进行了研究, 能实现0.5 mg·L-1级别的痕量检测; Nie[6]对2, 4-D等农药的THz吸收峰进行了特征提取和定量研究, 检出限可达5%。 这些研究大多专注于实验研究和定量测试, 或以单分子为模型进行理论计算并解析实验所得特征吸收峰的起源。
啶虫脒, 又称吡虫清, 乙虫眯, 是一种新型烟碱类杂环杀虫剂, 具有强内吸性和高杀虫活性, 对鳞翅目和缨翅目等害虫有高效杀灭作用。 分子式为C10H11ClN4, 化学名称为N'-[(6-氯-3-吡啶基)-甲基]-N-氰基-N'-甲基乙眯, 分子结构如图1所示。
 | 图1 标号的啶虫脒分子结构图Fig.1 The labeled molecular structure of acetamirid |
工作中利用THz-TDS技术获得了室温条件下啶虫脒在0.3~3.3 THz范围内的特征吸收谱, 并基于密度泛函理论分别对啶虫脒的孤立分子和晶胞进行建模、 结构优化和晶格动力学计算。 本研究对于揭示啶虫脒等有机农药分子在太赫兹波段特征吸收峰的形成机制, 获得这些分子结构与其太赫兹指纹谱的关系等, 具有重要的理论价值和实际意义。
实验所用的啶虫脒农药为无色结晶纯品, 购于Tokyo Chemical Industry Co., Ltd, 纯度大于99%, 使用之前未经进一步提纯。 将啶虫脒样品与高纯度聚乙烯粉末以1:10的比例混合后在研钵中充分研磨, 用精密电子天平称取150 mg放入FW4A压片机以8 MPa的压力压成直径13 mm, 厚度1.1 mm的圆形薄片。 样品片结构均匀, 前后表面光滑且平行。
光谱测量采用的THz-TDS实验装置是Tera View公司的TPS3000系统。 激光中心波长780 nm, 重复频率78 MHz, 利用光电导天线产生THz脉冲, 采用自由空间电光采样方法进行THz脉冲探测。 系统的光谱范围为0.05~4.0 THz, 峰值信噪比75 dB, 频谱分辨率2.0 GHz。
实验在室温293 K, 实验箱充满氮气的环境下进行, 样品信号及参考信号的时域光谱数据经傅里叶变换后得到频谱, 然后根据Duvillaret等提出的物理模型[7, 8], 通过式(1)和式(2)获得样品的折射率系数和吸收系数。
式(1)和式(2)中, ω 为角频率, c为真空中的光速, d为样品厚度, ϕ (ω )为样品信号与参考信号的相位差, ns(ω )为样品的折射率系数, α (ω )为样品的吸收系数, Es(ω )和Er(ω )分别为啶虫脒样品信号和参考信号经傅里叶变换后的频域信号。
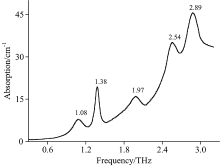
啶虫脒样品在0.3~3.3 THz范围内的吸收谱如图2所示。 从图中可以看出, 啶虫脒存在5个明显的特征吸收峰, 分别位于1.08, 1.38, 1.97, 2.54和2.89 THz, 强吸收峰出现在1.38和2.89 THz处, 另外三个吸收峰相对较弱。 吸收谱线随频率的增加呈上升趋势, 这是由于光散射和样品宽而无结构的吸收所致。 同时, 吸收峰的位置和强度还会受到系统噪声和基线漂移的影响, 这也是实验谱和模拟谱存在不规律偏差的原因之一。
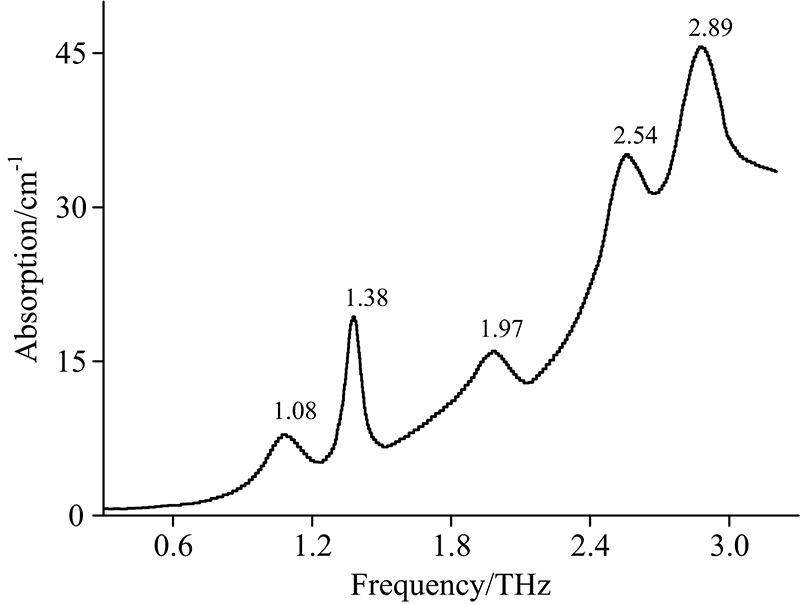 | 图2 啶虫脒的THz实验吸收谱Fig.2 The THz experimental spectra of acetamiprid |
为更好地理解啶虫脒太赫兹实验光谱产生的理论机理, 分别借助Gaussian09软件对孤立单分子模型、 Materials Studio8.0软件对晶胞模型进行了几何优化和振动频率计算。
首先使用Chemdraw获得啶虫脒单分子的结构式, 然后导入Chem3D中形成分子的立体结构并进行构象优化, 最后导入到GaussView中产生高斯计算的输入文件进而完成单分子理论模拟的建模。
单分子理论模拟计算在Gaussian09软件中进行, 采用基于密度泛函理论的B3LYP杂化泛函方法, 选取6-311G(d, p)基组进行几何优化, 并在相同水平上进行了振动频率计算。
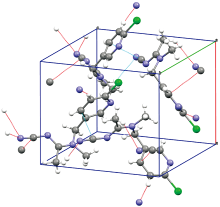
为探究氢键、 范德华力等分子间弱相互作用对太赫兹吸收谱的影响, 对啶虫脒晶体进行了理论模拟。 啶虫脒的晶胞模型[9]来自剑桥晶体结构数据库(Cambridge Crystallographic Data Centre, CCDC), 参考号: HANBAA。 该晶胞结构在室温293 K下通过X射线衍射技术获得并经过修正, 本文直接利用该模型进行结构优化和能量计算。 如图3所示, 啶虫脒晶胞属于正交晶系, 每个晶胞中包含有4个分子, 空间群为Pca21, 晶轴长度分别为: a=8.776 Å, b=11.780 Å, c=10.645 Å, 三条晶轴之间为正交关系: α =β =γ=90°。 图3中虚线所显示的分子间弱相互作用以C—H…N氢键为主, 同时此类氢键对晶格固定起主要作用。
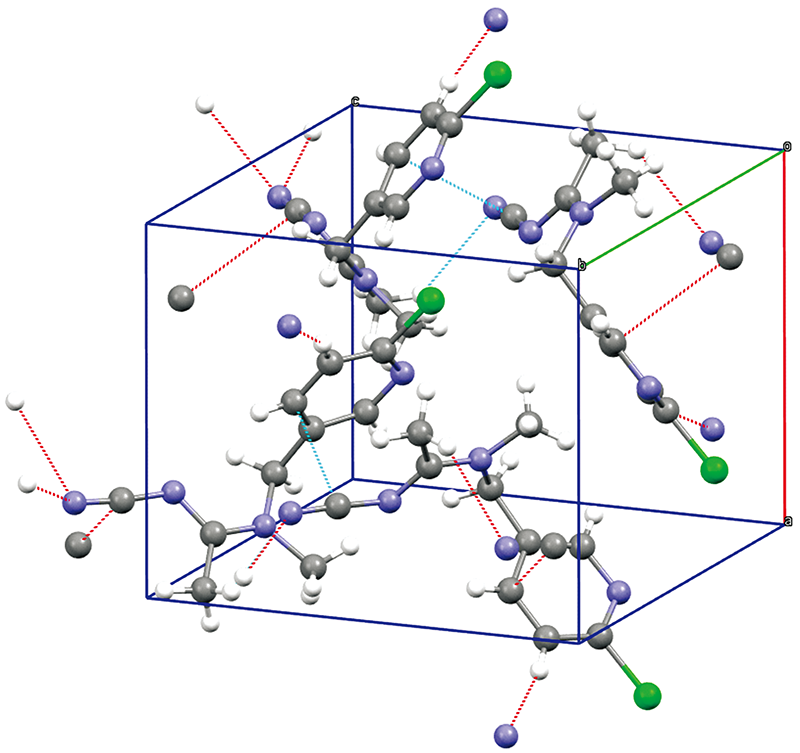 | 图3 啶虫脒的晶胞结构Fig.3 The Unit cell structure of acetamiprid |
晶胞的理论模拟在MS8.0的适合计算周期性结构的CASTEP[10]模块中进行。 采用基于平面波赝势的密度泛函理论和广义梯度近似(GGA)方法的PW91, PBE, PBEsol和WC四种交换相关泛函对晶胞模型进行几何优化和晶格动力学计算。 几何优化时采用BFGS (Broyden-Fletcher-Goldfarb-Shanno)拟牛顿算法中的线搜索(line search)方法进行。 在对原子位置几何优化的同时还对整个晶胞结构进行了优化。 几何优化的收敛精度设置为“ fine” , 对应的四个主要参数收敛阈值: 能量(Energy)为10-5 eV·atom-1, 受力(Max.force)为0.03 eV·Å-1, 压力(Max.stress)为0.05 GPa, 位移(Max.displacement)为10-3 Å。 平面波截断能为1 000 eV, 选择模守恒赝势基组, 使用Γ 点计算振动频率[11]。
表1列出了啶虫脒分子的X射线衍射(XRD)实验测量结构参数(键长、 键角)、 单分子理论模拟结构参数和晶胞理论模拟结构参数, 给出了各模拟值与实验值的均方根偏差RMSD[12]。
式(3)中, xcal和xexp分别是结构参数的模拟值和实验值。
RMSD值直观地表示了不同理论方法模拟的键长、 键角参数与XRD实验值之间的偏离程度。 由表1可看出, 模拟值与实验值基本相符, 绝大部分的键长模拟值与实验值间的差别仅为0.002 nm, 键角仅有1°的偏差。
| 表1 啶虫脒分子的理论键长、 键角及其相对于实验数据的RMSD值 Table 1 Calculated bond lengths (Å), bond angles (°) and their RMSD values of acetamiprid compared to experimental X-ray values |
图4(a, b)分别给出了键长、 键角的各理论模拟值与实验值之间偏差值的变化图, 更清晰地展现了啶虫脒分子理论模拟结构与实验结构的差异。 在键长差值的对比中, 孤立分子和固态晶胞的计算结果与实验值间的偏差总体不大, 表现出了较好的一致性, 其中基于B3LYP方法获得的单分子键长的RMSD为0.017 6, 相较于基于四种固态GGA泛函方法的晶胞中分子的键长RMSD, 偏差值更小。 但是, 从图4(a)所示的键长差值变化可以看出, 气态孤立分子状态下, 其键长在正负两个方向上都出现了较大变化, 在稳定性方面没有固体形态好。 B3LYP方法模拟结果的较大偏差出现在N2-C8处, 键长偏差为0.03 Å; 在N3-C8和N1-C5处还出现了负偏差, 差值分别为-0.003和-0.002 Å。 由图4(a)还可以看出, PW91, PBE, PBEsol和WC四种固态GGA泛函对键长的优化结果比较一致, 变化方向也是趋于相同, 其中PBE的计算结果始终处于四种固态计算结果的均值位置, 显示出了更好的稳定性。
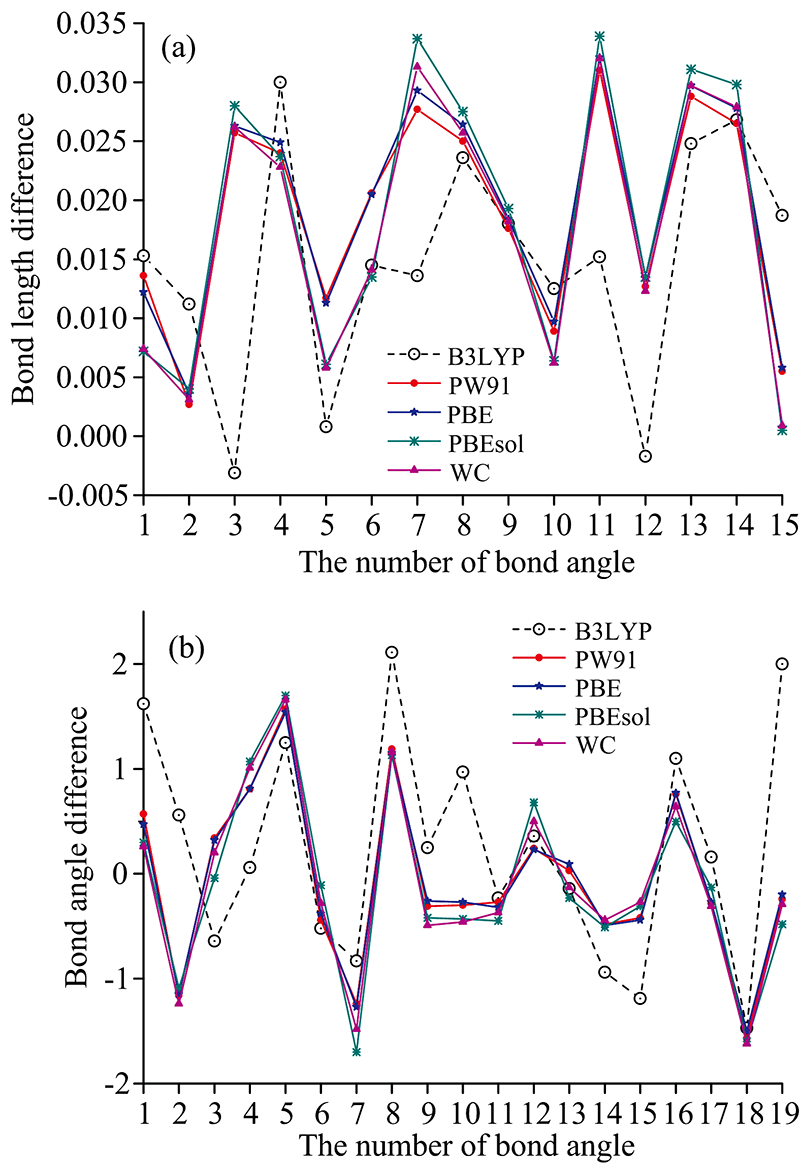 | 图4 啶虫脒分子的键长(a)、 键角(b)差值变化图Fig.4 Bond length (a) and bond angle (b) differences between calculations and X-Ray diffraction results |
由图4(b)可以看出, 四种固态GGA泛函的键角模拟值与实验值偏差的变化趋势表现出了很好的一致性。 相比之下, 基于B3LYP的单分子的键角模拟结果表现出了明显差别。 单分子的键角RMSD值为1.057, 远大于四种固态GGA算法所得的结果。 这种差异说明单分子的模拟结果与实验值差异较大, 因此用单分子的计算谱来模拟实验谱, 必定存在较大的偏差。 PBE泛函的计算结果与实验值之间的偏差最小, RMSD值为0.776, 这也在一定程度上印证了PBE理论计算谱与实验谱更为接近的内在机理。
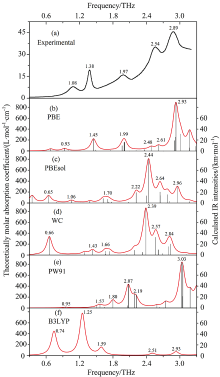
图5给出了啶虫脒的基于孤立分子和晶胞模型的模拟仿真谱与实验吸收谱的对比, 实验谱图5(a)由THz-TDS获得, 图5(b—f)所示的模拟谱利用Multiwfn3.0软件[13]绘制, 利用洛伦兹函数并以半高宽(full width at half maximum, FWHM) 4 cm-1对红外振动强度(图中竖线)进行拟合得到。 由图5(f)可知基于密度泛函理论, 使用B3LYP泛函和6-311G(d, p)基组对单分子模型进行的计算在0.3~3.3 THz范围内产生了5个简正模式, 这5个简正模式与实验所得到的特征吸收峰无论在峰位、 强度和波形上都完全不匹配。 这种对孤立单分子的计算模拟只能反映常温气相状态下分子内的振动模式, 不能够反映分子间的相互作用, 与实验中啶虫脒的固态形态存在显著差异, 因而不能用单分子模型对啶虫脒太赫兹谱进行模拟计算和光谱解析。
 | 图5 啶虫脒实验吸收谱(a)与理论模拟谱(b, c, d, e, f)的对比Fig.5 The comparison of experimental spectrum (a) of acetamiprid and its simulated spectra (b, c, d, e, f) |
为更准确地模拟并解释啶虫脒的太赫兹实验吸收谱, 同时为了验证GGA密度泛函计算农药晶体的性能, 利用PBE, PBEsol, WC和PW91四种泛函对啶虫脒晶胞模型进行了几何优化和晶格动力学计算, 计算结果均无虚频出现, 证明优化得到的结构是一个稳定的形态。 由图5(b, c)可知在0.3~3.3 THz范围内, PBE和WC产生了8个简正模式, 由图5(b, e, f)可知看出PBEsol和PW91产生了9个简正模式。 这些简正模式的频率位置和振动强度对实验吸收谱的模式指认都非常重要。 由图5可知, 从多数简正模式的频率位置、 振动强度以及拟合谱线形态上看, PBEsol和WC两种泛函的模拟结果非常相近, 但与实验吸收谱却不吻合。 PW91和PBE泛函的模拟结果与实验吸收谱在频率位置、 振动强度以及谱线形状上比较接近, 这可能是因为两者优化后的结构参数比PBEsol和WC的优化结果更接近于晶体本身。 PBE比PW91更好地再现了实验所测得的特征峰, 分析认为, 一方面是因为PBE的键角RMSD比PW91更小, 另一方面是因为PBE泛函从理论上改进了PW91, 可以更精确地描述原子、 分子、 晶体的自旋局域密度, 进而可以更精确地描述体系的能量。
由上述分析可知, PBE泛函的模拟结果与实验吸收峰最为接近。 因此用PBE的简正模式对相应的实验特征峰进行解析更为准确。
表2列出了啶虫脒在0.3~3.3 THz范围内的实验吸收峰及其对应的PBE模拟吸收峰的位置和强度, 并给出了振动模式归属。 分子晶体的振动模式分为外振动和内振动, 外振动即分子间振动反映了晶体结构及其对称性, 内振动主要是指分子内的变形振动。 外振动的频率通常低于内振动的频率。 PBE理论预测的啶虫脒分子晶体的所有振动模式都是外振动模式。
| 表2 啶虫脒吸收峰振动模式归属 Table 2 Vibrational modes Assignment of corresponding peaks |
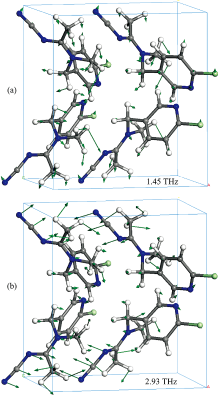
图6(a, b)分别给出了1.45和2.93 THz处两个强度最大的PBE泛函简正振动模式的原子位移矢量图。 直观地展示了分子晶体中复杂的振动现象, 这些振动主要还是由晶体内以C—H…N氢键为主的分子间弱相互作用带动的骨架振动和基团的整体振动。
 | 图6 两个最强简正模式1.45 THz (a), 2.93 THz (b)的PBE泛函模拟位移矢量Fig.6 The simulated displacement vectors for the most intense mode at 1.45 THz (a), 2.93 THz (b) of the PBE functional |
利用THz-TDS技术获取了啶虫脒晶体在0.3~3.3 THz频段的特征吸收谱。 使用五种基于密度泛函理论的计算方法对实验THz谱进行解析, 发现基于晶胞模型的量子化学计算由于考虑了氢键、 范德华力以及晶格振动等分子间相互作用, 比单分子模型的模拟计算更准确的模拟有机农药分子晶体在太赫兹频段的吸收特征。 通过比较四种GGA泛函, 发现基于PBE泛函的计算结果中, 键长、 键角等分子的结构数据与X射线衍射实验数据的一致性较好, 其仿真谱与实验谱也最为吻合。 依据PBE泛函的计算结果对太赫兹特征吸收峰对应的振动模式进行了归属。 研究表明, 啶虫脒晶体在THz频段的光谱特性主要来源于由C—H…N分子间氢键主导的分子基团的整体振动以及由此引发的分子内集体振动模式。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|