{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
竹基纤维素Ⅱ氢键结合的近红外光谱研究
[董沛杰1  , 张文博
, 张文博1, * , 陆薇2 ]
, 张文博, 陆薇|
|


作者简介: 董沛杰, 1996年生, 北京林业大学材料科学与技术学院硕士研究生 e-mail: 1278616670@qq.com
纤维素是一种可再生天然亲水性高聚物, 其庞大的氢键网格形成多种不同的晶体结构形式。 在其五种结晶变体(纤维素Ⅰ, Ⅱ, Ⅲ, Ⅳ和Ⅹ)中, 纤维素Ⅱ由纤维素Ⅰ(天然纤维素)经再生或丝光化处理获得, 是表面自由能最低、 性能最稳定的纤维素, 这主要归因于纤维素Ⅱ具有与纤维素Ⅰ晶型平行链结构相反的反平行链结构, 且相比于纤维素Ⅰ有附加的分子间氢键。 基于近红外光谱(NIRS)对含氢基团的敏感性及纤维素的结晶结构中有大量氢键, 使通过NIRS定性检测、 定量评价纤维素的结晶结构成为可能。 目前, 用NIRS对纤维素结晶变体氢键结合的研究甚少, 针对竹材纤维素Ⅱ及其衍生材料氢键结合的研究国内外尚未见相关报道。 用竹材制备纤维素Ⅰ, 经丝光化处理得到竹基纤维素Ⅱ, 通过NIRS研究其氢键结合状况, 结果与竹粉及竹基纤维素Ⅰ相比较。 此外, 研究还通过NIRS对竹粉及竹基纤维素的结晶度做了定量评价。 结果表明: (1)在无定形区, 竹基纤维素Ⅰ、 Ⅱ和竹粉相比光谱差异不大, 氢键结合只有量的变化, 而无质的差异; (2)在半结晶区, 与竹粉相比, 竹基纤维素Ⅰ晶型结构保持不变, 而竹基纤维素Ⅱ形成双峰; (3)在纤维素结晶区的近红外谱带范围内, 反映竹基纤维素Ⅰ结晶表面纤维素分子内氢键O2—H2…O6的强氢键结合的羟基伸缩振动的一次倍频吸收峰由6 292 cm-1向高波数转移到6 354 cm-1, 该处与竹基纤维素Ⅱ形成的强氢键结合的分子间氢键O2—H2…O2反平行构造相对应; (4)NIRS预测的结晶度与XRD分析结果有良好相关性。 上述结果表明, 纤维素结晶区内的氢键结合在近红外特征谱带出现转移且在半结晶区形成双峰, 是区别竹基纤维素Ⅱ和Ⅰ的主要特征。 研究也表明NIRS对探讨纤维素多种变体的氢键结合及结晶度预测是可靠的。
Cellulose is a renewable natural hydrophilic polymer, and its huge hydrogen bond grid forms a variety of different crystal structures. There are five crystalline variants of cellulose (cellulose Ⅰ, Ⅱ, Ⅲ, Ⅳ and Ⅹ), of which cellulose Ⅱ is formed from cellulose Ⅰ (natural cellulose) after regeneration or mercerization, is the lowest surface free energy and the most stable performance among the five crystal varieties, mainly due to the antiparallel chain structure of cellulose Ⅱ, which is opposite to the parallel chain structure of cellulose I, and has additional intermolecular hydrogen bonds compared with cellulose I. Therefore, in view of the sensitivity of near-infrared spectroscopy (NIRS) to the hydrogen-containing group, and the crystalline structure of cellulose contains a large number of hydrogen bonds, this makes it possible for NIRS to analyze the degree of hydrogen bond destruction of cellulose by hydrogen containing functional groups, and to detect and quantitatively evaluate the crystalline structure of cellulose qualitatively. So far, there are very few studies on the hydrogen bonding of cellulose crystal variants, and the hydrogen bonding of bamboo cellulose Ⅱ and its derivatives has not been reported at home and abroad. In the study, cellulose Ⅰ was prepared from bamboo, and bamboo-based cellulose Ⅱ was obtained through mercerization, which NIRS investigated hydrogen bonds, the results were compared with bamboo powder and bamboo-based cellulose Ⅰ. Besides, the crystallinity of bamboo powder and bamboo-based cellulose was quantitatively evaluated by NIRS. The results can be drawn as follows: (1) the differences of NIRS among bamboo powder, bamboo-based cellulose Ⅰ and Ⅱ varied little, hydrogen bonding were quantitatively remarkable, but were qualitatively slight in the amorphous region; (2) compared with bamboo powder, the crystal structure of bamboo-based cellulose I remained unchanged, while bamboo-based cellulose Ⅱ occurred two absorbance peaks in the semi-crystalline region; (3) in the crystalline region a strong hydrogen bonding absorbance peak reflected the first overtone of hydroxyl group stretching vibration was observed at the wavenumber of 6 292 cm-1 assigned to the intermolecular bond of O2—H2…O6 of cellulose Ⅰ, which shifted to 6 354 cm-1 for bamboo-based cellulose Ⅱ. We deduced the absorbance peak in cellulose Ⅱ was assigned to the intermolecular bond of O2—H2…O2 due to anti-parallel structure of cellulose confirmation; (4) a good correlation among crystallinity was obtained by NIRS with the results of XRD analysis. The above research shows that the hydrogen bonding in the crystalline region of cellulose shifts in the near-infrared characteristic band and forms double peaks in the semi-crystalline region, which were the main characteristics of bamboo-based cellulose Ⅱ different from bamboo-based cellulose Ⅰ. Simultaneously, it is feasible to use NIRS to study the hydrogen bonding of various celluloses and predict their crystallinity.
纤维素Ⅰ 和Ⅱ 是纤维素两种最主要的同质多晶体, 纤维素Ⅰ 属于平行链结构, 纤维素Ⅱ 则是反平行链结构[1], 且相比纤维素Ⅰ 有附加的分子间氢键。 结构差异导致纤维素Ⅱ 化学稳定性更好, 意味着在做纳米纤维素填料增强剂开发纤维素纳米复合材料时, 纤维素Ⅱ 的分散性和兼容性更好。 有研究表明, 纤维素Ⅱ 纳米纤维制备的增强薄膜材料具有高韧性、 低热膨胀系数及高热稳定性, 主要归因于纤维素Ⅱ 的结晶结构中氢键结合更稳固[2]。 本文用竹粉制备纤维素Ⅰ 和Ⅱ , 通过近红外光谱(near-infrared spectroscopy, NIRS)研究其氢键结合状况。 关于用光谱研究聚合物晶型及结构的报道很多, 如红外光谱对纤维素Ⅰ 和Ⅱ 的结构分析表明, 其在指纹区无基团振动差异, 仅在羟基吸收区3 488和3 445 cm-1处有两个微弱吸收峰[3]。 然而关于纤维素结晶变体氢键结合的NIRS研究则很少。 NIRS反映大量含氢基团的振动信息, 适用于木质生物质等亲水性材料的组成结构研究, 并在以木质材料为主的高分子材料结构研究中应用广泛。 Horikawa[4]通过NIRS分析多种植物源纤维素的结晶构造并建立分类预测模型。 结果表明, 通过NIRS吸收的谱带转移能够简便获得α 和β -纤维素结构上的差异信息, NIRS能定性识别不同植物源纤维素。 Inagaki[5]通过NIRS结合XRD技术定量评价水热处理日本扁柏与古建木构件结晶结构的差异, 两种技术分析结果有良好相关性, 表明NIRS可定量评价木材纤维素结晶度变化。 本研究用NIRS对制备的两种晶型的纤维素从氢键结合的角度进行定性分析国内外尚未见相关报道, 同时结合XRD的分析结果, 探讨NIRS定量评价和预测纤维素结晶变体结晶度的可能性。
原材料为3年生以上的毛竹(Phyllostachys heterocycla cv. Pubescens), 去除竹青竹黄后磨粉过60目筛。 所用试剂氢氧化钠(NaOH)、 氢氧化钾(KOH)、 亚氯酸钠(NaClO2)和冰醋酸(CH3COOH)(麦克林, 中国)均为分析纯级别。
1.2.1 制备竹基纤维素Ⅰ (纯化纤维素)
步骤一, 取2 g竹粉放入蒸馏水中, 加0.75 g NaClO2, 滴入CH3COOH, 使pH值保持在4~5, 置于75 ℃恒温水浴锅中1 h, 并重复5次直至样品变白, 抽滤、 洗涤至中性以脱除木质素得到综纤维素; 步骤二, 在室温下用5% KOH溶液处理综纤维素12 h, 再置于90 ℃恒温水浴锅中2 h, 抽滤、 洗涤至中性以去除大部分半纤维素; 最后, 将样品进一步纯化, 同步骤一。
1.2.2 制备竹基纤维素Ⅱ (丝光纤维素)
按1.2.1方法制得竹基纤维素Ⅰ , 并用17.5 Wt% NaOH于室温下处理12 h, 抽滤、 洗涤至中性, 烘干后备用。
采用Ultima Ⅳ 型X射线衍射仪(理学, 日本), 电压40 kV、 电流40 mA, 用铜靶, 扫描角度5° ~40° , 扫描速度2° · min-1。 所得XRD原始数据经MDI Jade软件分析(扣除背景及峰形拟合等), 再用Origin软件绘图(平滑处理及相关标注等)。 每种样品检测5个, 结果取其平均值。 相对结晶度CrI的计算采用Segal[6]等提出的峰强度法, 其计算公式如式(1)
其中, I002(2θ =22.7° )为002面峰强度, 即纤维素结晶区的衍射强度, IAM(2θ =18° )为无定形区的衍射强度。
采用MPA型近红外光谱仪(Bruker, 德国), 扫描波数12 000~4 000 cm-1, 分辨率8 cm-1, 扫描次数32次。 采用漫反射模式, 每次扫描前晃动样品以保证样品多个部位均被扫描, 每个位点自动扫描32次, 每次约10 s, 取光谱平均值。 NIRS原始数据通过Origin软件进行数据处理(基线校正及峰面积计算等)。 每种试样平行试验进行6次, 结果取其平均值。
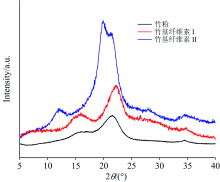
样品XRD结果如图1, 竹粉的衍射峰主要出现在约2θ =16.02° , 22.2° 处, 显示天然纤维素的典型结晶特征[7]。 经酸性亚氯酸钠法纯化处理后, 样品整体趋势上与竹粉结晶构型相似, 在2θ =14.9° , 16.5° , 22.26° 处出现衍射峰, 是纤维素Ⅰ 的典型结晶特征, 说明经纯化处理脱木素和半纤维素后纤维素的结晶构型并未改变, 仍保持纤维素Ⅰ 的结晶结构。 而经17.5 Wt% NaOH丝光处理后, 衍射峰主要出现在2θ =12.1° , 20.1° , 21.5° 处, 表现出竹基纤维素Ⅱ 和I结晶特征的差异, 与French[7]对Ⅰ 型和Ⅱ 型纤维素结晶构型的研究结果一致。 XRD结果表明, 竹粉及竹基纤维素Ⅰ 均保持天然纤维素的晶型结构。 而经17.5 Wt% NaOH丝光后, 纤维素结晶构型由Ⅰ 型转变为Ⅱ 型, 成功制备出了竹基纤维素Ⅱ 。
 | 图1 竹粉、 竹基纤维素Ⅰ 及Ⅱ 的X射线衍射图Fig.1 The XRD patterns of Bamboo powder, Bamboo-based cellulose Ⅰ and Ⅱ |
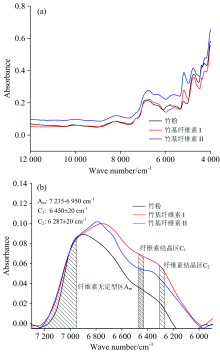
如图2(a)所示, 从高波数到低波数, 三种样品NIRS光谱趋势大致相同, 差别主要体现在7 200~6 000和5 400~4 200 cm-1区间。 其中, 7 200~6 000 cm-1区间主要反映纤维素上羟基伸缩振动的一次倍频[8], 在图2(a)中出现一个较强吸收峰; 而5 400~4 200 cm-1区间主要反映羟基伸缩振动与变形振动的合频, 及羟基伸缩振动与亚甲基各种振动的合频[8], 在图2(a)中出现了两个明显吸收峰。 因此, 主要对上述两个波数区间进行分析与讨论。
 | 图2 竹粉、 竹基纤维素Ⅰ 及Ⅱ 的NIRS原始谱图(a) 及7 200~6 000 cm-1放大图(b)Fig.2 The NIRS original spectra (a) of Bamboo powder, Bamboo-based cellulose Ⅰ and Ⅱ and their partial spectra at the wavenumber range of 7 200~6 000 cm-1(b) |
Tsuchikawa和Siesler[9]根据木质高分子材料中不同分子结构多糖中的羟基结合状态, 把NIRS在波数大于6 950 cm-1归为无定形区, (6718± 20) cm-1为半结晶区, 而结晶区在(6 450± 20)和(6 287± 20) cm-1范围。 按这一划分对NIRS原始谱图进行基线校正及平滑处理, 获得其在7 200~6 000 cm-1的局部放大图, 如图2(b)。 图2(b)中, 在波数大于6 950 cm-1范围内, 三种样品谱线覆盖区域差别不大, 约6 900 cm-1处开始有差别: 竹粉吸收峰强度减小, 两种纤维素吸收峰强度以同样趋势增加, 约6 800 cm-1处达到最大吸收强度。 在6 450和6 287 cm-1附近两个结晶区域内, 竹基纤维素Ⅱ 的光谱吸收强度明显低于竹基纤维素Ⅰ 。 图2(b)表明, 在无定形区, 氢键结合状态不受竹材化学组分变化的影响, 而在半结晶和结晶区, NIRS吸收强度则出现明显的差异。
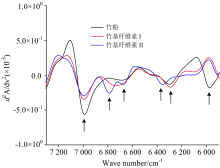
三种试样在近红外光谱7 200~6 000 cm-1区间吸收峰的变化, 通过二阶导数处理结果如图3。 三种试样反映的特征吸收峰如图3中箭头所示。 从高波数到低波数, 6 990 cm-1是纤维素非晶区自由羟基及羟基弱氢键伸缩振动的一次倍频[8]。 通过脱化学组分处理, 随着纤维素Ⅰ 和Ⅱ 中半纤维素的减少, 吸湿性降低, 吸收峰强度明显降低, 但两者的吸收峰位置及吸光度差别不大, 表明在非结晶区, 两种晶型结构的竹基纤维素与竹粉中自由羟基形成的氢键结合没有本质差别。以6 700 cm-1为中心出现一个较弱吸收峰, 归属于纤维素半结晶区羟基伸缩振动的一次倍频[10], 纤维素Ⅱ 在6 790和6 674 cm-1处出现一强一弱两个峰, 反映了竹基纤维素Ⅱ 与Ⅰ 晶型构造的差异。 在约6 292 cm-1出现的吸收峰, 反映纤维素结晶区羟基伸缩振动的一次倍频, 为纤维素分子内氢键O2— H2…O6的强氢键结合[11]。 从该处可看出, 竹粉和竹基纤维素Ⅰ 吸收峰位置没有变化, 表明两者结晶构造相同, 而竹基纤维素Ⅱ 向高波数方向转移到6 354 cm-1附近, 表明竹基纤维素Ⅱ 结晶区表面羟基形成的分子间强氢键结合发生转移。 因此, 6 354 cm-1附近可作判别竹基纤维素Ⅰ 与Ⅱ 的特征吸收峰。 基于纤维素Ⅱ 的反平行结构, 推测该处为羟基形成的分子内氢键O2— H2…O2的强氢键结合。 此外, 在5 974 cm-1处反映木质素芳环骨架上CH基团伸缩振动一次倍频峰[12], 该处竹粉检测到一个明显吸收峰, 而两种纤维素的吸收峰则消失, 表明竹基纤维素Ⅰ 及Ⅱ 中木质素已经脱除。
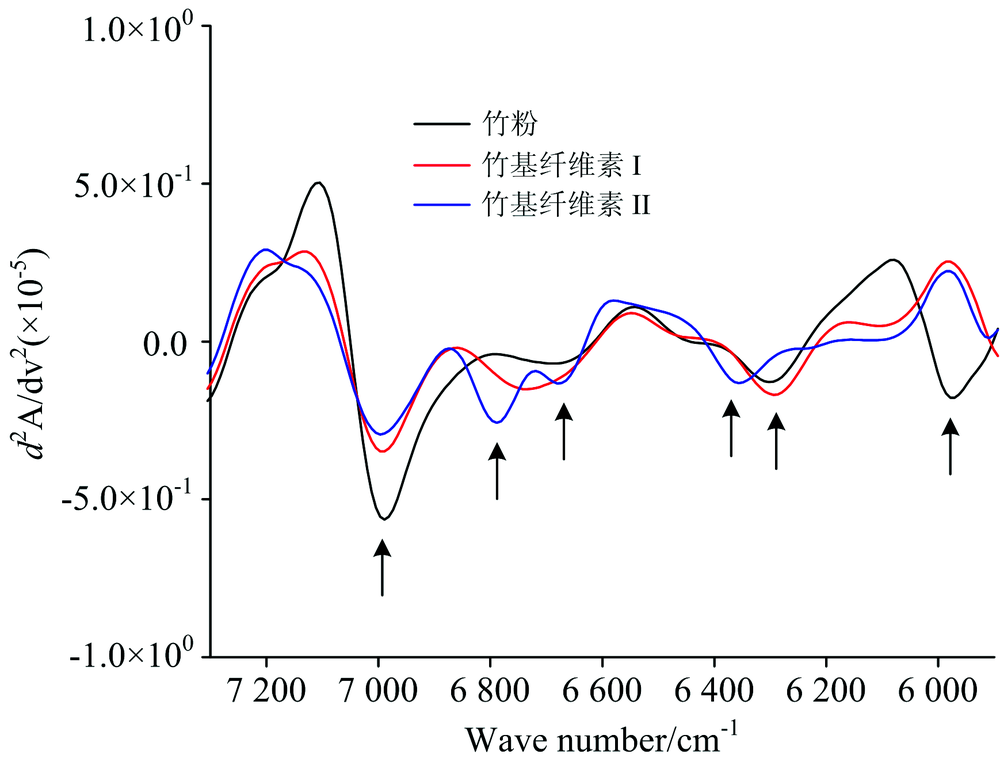 | 图3 竹粉、 竹基纤维素Ⅰ 及Ⅱ 在7 200~6 000 cm-1范围内的NIRS二阶导数图Fig.3 Second-derivative spectra of Bamboo powder, Bamboo-based cellulose Ⅰ and Ⅱ at the wavenumber range of 7 200~6 000 cm-1 |
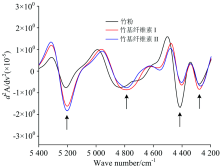
在近红外光谱5 400~4 200 cm-1区间, 三种试样的光谱变化经二阶导数处理, 特征吸收峰如图4。 从左往右箭头所示, 可明显观察到5 200, 4 795, 4 404和4 289 cm-1处的4个吸收峰。 其中, 5 200 cm-1处归属于纤维素自由羟基非对称伸缩振动和变形振动的合频[13], 与样品中自由水有关。 4 795 cm-1处归属于纤维素羟基伸缩振动和C— H基团变形振动的合频, 三种试样在该处吸收峰差异不大, 均表现为较宽峰形。 4 404 cm-1处归属于纤维素和半纤维素中羟基伸缩振动和C— H2基团变形振动的合频[14], 与竹粉相比, 两种晶型的竹基纤维素在该处的吸收峰强度明显减弱, 表明两种竹基纤维素中半纤维素的相对含量减少。 此外, 4 289 cm-1处归属于纤维素和半纤维素上C— H基团伸缩振动及其变形振动的合频[15], 三种试样在该处没有差异。 在红外光谱指纹区(1 300~400 cm-1)内, 纤维素Ⅰ 和纤维素Ⅱ 有机基团结构无差别, 因此通过红外光谱识别纤维素Ⅰ 和纤维素Ⅱ 存在困难, 但通过NIRS能明确地观察与辨别纤维素Ⅰ 和纤维素Ⅱ 的差异。
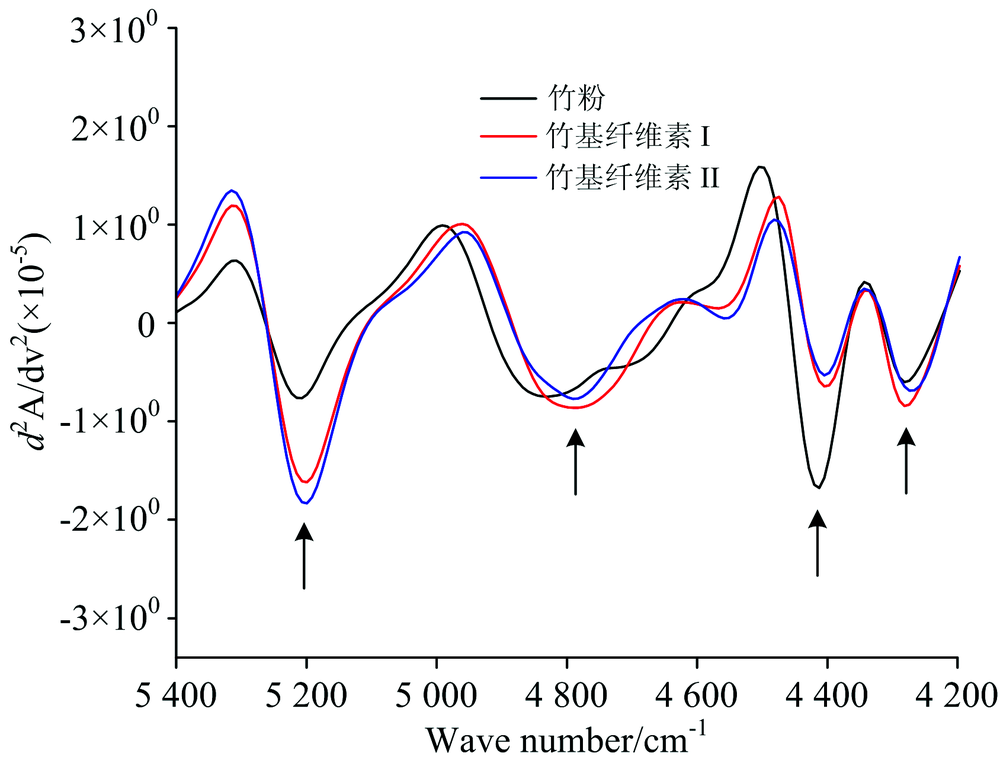 | 图4 竹粉、 竹基纤维素Ⅰ 及Ⅱ 在5 400~4 200 cm-1范围内的NIRS二阶导数图Fig.4 Second-derivative spectra of Bamboo powder, Bamboo-based cellulose Ⅰ and Ⅱ at the wavenumber range of 5 400~4 200 cm-1 |
由于NIRS在波数6 450和6 287 cm-1附近反映纤维素结晶区表面羟基形成的氢键结合吸收峰。 据这一推论, 结合XRD对结晶度的分析, 对NIRS法估算纤维素结晶度进行了评价。 由NIRS法计算样品结晶度的公式如式(2)[5]
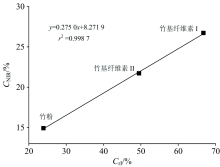
其中, A(C1), A(C2)和A(Am)分别表示纤维素样品结晶区和无定形区范围内的积分面积。 通过图2(b)中确定的波数区间7 235~6 950, (6 450± 20)和(6 287± 20) cm-1分别计算C1, C2及Am区域的积分面积。 图5表明, NIRS和XRD两种方法计算的样品结晶度之间的相关系数(r2)达到0.99, 所得结果有良好的相关性。 该结果也表明了NIRS技术快速准确预测纤维素样品结晶度的可行性。
 | 图5 XRD法结晶度与NIRS法相对结晶度的线性拟合Fig.5 Relationship between the degree of crystallinity calculated from XRD (CrI) and from NIR spectra (CNIR) |
使用NIRS对竹基纤维素Ⅱ 的氢键结合状况进行了研究, 将实验结果与未处理竹粉及竹基纤维素Ⅰ 进行比较。 研究表明: (1)在无定形区, 竹基纤维素Ⅰ , Ⅱ 和竹粉相比光谱变化不大, 氢键结合只有量的变化, 而无质的差异; (2)在半结晶区, 与竹粉相比, 竹基纤维素Ⅰ 晶型结构保持不变, 而竹基纤维素Ⅱ 形成双峰; (3)在纤维素结晶区的近红外谱带范围内, 反映竹基纤维素Ⅰ 结晶表面纤维素分子内氢键O2— H2…O6强氢键结合的羟基伸缩振动吸收峰由6 292 cm-1向高波数转移到6 354 cm-1, 该处与竹基纤维素Ⅱ 形成的强氢键结合的分子间氢键O2— H2…O2反平行构造相对应; (4)NIRS对纤维素结晶度的定量分析结果与XRD分析结果有良好相关性。 两种晶型纤维素结晶区内的氢键结合在近红外特征谱带出现转移且在半结晶区形成双峰, 表明竹基纤维素Ⅰ 和Ⅱ 形成了不同的氢键结合, 决定两者结晶构造的差异, 从而对其物理、 化学性质产生影响。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|