
{kind=link}
{kind=link}
X射线光谱技术在生物与生态环境中的应用进展
[柳检1  , 劳昌玲
, 劳昌玲2 , 袁静3 , 孙梦荷4 , 罗立强5 , 沈亚婷5, * ]
, 劳昌玲]
|
|


作者简介: 柳 检, 1990年生, 浙江大学环境与资源学院在站博士后 e-mail: liujian120129@126.com
环境样品中元素的浓度、 空间分布和赋存形态等是认识元素的生物功能和环境行为的关键。 本文对近年来X射线光谱技术在生物与生态环境中的应用研究进展进行了评述, 发现X射线荧光光谱技术可以提供活体植物中元素迁移与分布的定量数据, 且微区X射线荧光光谱和X射线吸收谱技术的联用可深入认识生物与元素的相互作用, 尤其是生物对元素的吸收、 转运、 贮存和细胞解毒机制, 同时也能够揭示典型环境样品中元素的来源、 演化和归趋等环境行为。 然而, 由于生物和环境样品基质的复杂性和多样性, 仍存在一些技术难点与挑战, 如X射线荧光自吸收效应的克服、 低丰度(5%~10%)的元素形态的准确鉴定, 以及对活体细胞中短暂的元素氧化还原反应的捕捉等。
The concentration, spatial distribution and speciation of elements in environmental matrices are the key to understanding their biological functions and environmental behaviors. This paper aims to review the recent applications and challenges of X-ray spectrometry in biology and ecological environment. It is demonstrated that X-ray fluorescence spectroscopy analysis can provide quantitative data on the translocation and distribution of elements in living plants. Micro-X-ray fluorescence and X-ray absorption spectroscopy are unique in providing in situ information, and both help to understand the interaction between organisms and elements, especially the uptake, transport, accumulation and detoxification mechanism of elements in organisms. Meanwhile, both are also used to reveal the environmental behaviors such as the source, evolution and fate of elements in typical environmental samples. However, due to the complexity and diversity of biological and environmental matrices, there are still some technical difficulties and challenges, such as overcoming the self-absorption effect of X-ray fluorescence, accurately identifying the low-abundance (5%~10%) elemental species, and rapidly arresting the transient redox reactions of elements in living cells.
生物、 土壤、 海洋沉积物和大气颗粒物等是环境中化学元素的重要载体, 研究各载体中元素的浓度、 空间分布和原子配位结构等是揭示元素迁移、 转化和富集的重要前提。 X射线光谱技术(XRS)具有制样简单、 固体进样、 非破坏和多元素分析等技术特点, 且随着X射线光管、 多层单色仪、 探测器等关键技术元件的不断更新, 以及基体校正方法、 计算机技术的发展, 在一定程度上提高了X射线荧光光谱(XRF)技术对大多数元素的灵敏度(μ g· g-1级)[1], 这极大的促进了XRF技术在生物、 土壤和大气颗粒物等样品分析中的广泛应用, 从而推动了生物地球化学和生态环境事业的发展。
微区X射线荧光光谱(μ -XRF)和X射线吸收谱(XAS)技术作为X射线光谱技术的重要分支, 也是生态环境样品中元素分析的重要手段和支撑技术。 μ -XRF技术能够在微米、 亚微米, 甚至纳米水平上深入到微小环境样品内部(如生物细胞), 进行深层次的矿物学、 生态学和生物学研究, 通过获取样品中元素的原位分布信息, 从而揭示物质的形成条件、 元素的动态分布及元素间的相互作用机理等[1]。 事实上, μ -XRF并不是一种独立的技术, 它常与其他同步辐射技术(如XAS和μ -XRD)互相补充(图1), 以提供基于元素空间分布图中感兴趣点位的价态、 原子配位结构和官能团构效等[2]。 XAS技术包括X射线近边吸收结构谱(XANES)和扩展边X射线吸收精细结构谱(EXAFS), 两者均可原位获取元素的形态信息, 且不受样品结晶度的限制[3]。
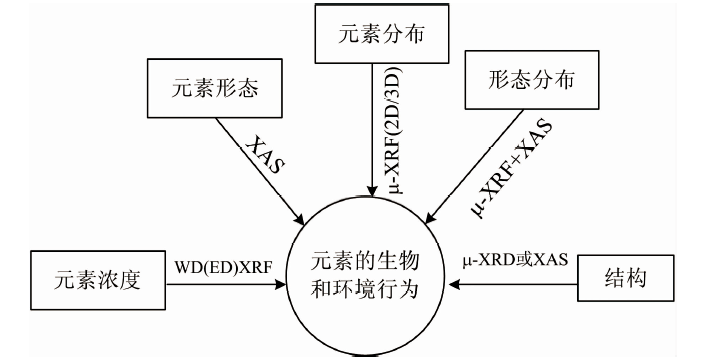 | 图1 X射线光谱技术在元素的生物和环境行为研究中的应用Fig.1 Application of X-ray spectroscopy in the study of biological and environmental behavior of elements |
X射线光谱技术的原位、 无损分析特点, 使其在生物与生态环境领域具有广泛的应用前景, 尤其是μ -XRF和XAS技术的结合。 本文对近年来X射线光谱技术在生物与生态环境领域中的应用研究进展进行了评述。 首先综述了μ -XRF和XAS技术在生物与元素相互作用研究中的应用现状与进展, 主要涉及两种技术在植物、 微生物、 动物和人体组织对元素吸收、 转运、 贮存、 解毒和生物代谢过程研究中的应用; 其次评述了μ -XRF, XAS和μ -XRD技术在环境领域的应用现状与进展, 包括大气系统及大气颗粒物、 海洋系统、 土壤与沉积物体系中元素的组成、 迁移和转化过程研究中的应用; 最后探讨了X射线光谱技术在该领域应用中还存在的技术难点和挑战, 以期为今后更好地推广该技术提供参考。
植物是生态系统的第一个营养层级, 研究植物中元素的浓度、 分布特征和赋存形态是衡量元素在生物链中的传递效率, 以及评估其环境风险的重要依据。 近年来, XRF和XAS技术被广泛应用于植物中元素的(半)定量分析、 微区分布和形态转化研究, 尤其是在微米甚至纳米尺度下, 获得活体植物中元素的生物环境信息[4]。
XRF技术具有前处理简单和原位无损分析的特点, 在植物样品的元素定量分析中独具优势[5]。 如结合波长色散(WD)和能量色散(ED)XRF分析药用植物中主、 微量元素, 其前处理过程仅需将植物粉末样品与石蜡混合(质量比10:1), 于20 t压力下压制成直径为40 mm样片后, 直接用于WD/ED-XRF测试, 光谱数据应用基本参数法计算, 即可获得样品中17种元素的浓度[6]。 也有学者未使用粘结剂, 直接对竹笋的粉末样品进行压片后, 运用WDXRF测试样品中主微量元素的浓度[7]。 另有学者在利用同步辐射X射线光谱技术对Pb的定量分析中, 发现Pb Lα 特征峰的强度与康普顿峰的强度之比与菜豆样品中Pb的浓度显著正相关(R2=0.994 1), 同时利用不同Pb浓度的菜豆茎样品建立校准曲线, 结果显示: 对于Pb 浓度高于90 μ g· g-1时, 其与ICP-OES测试结果的相对误差为1%~9%, 而低于90 μ g· g-1时, 两者的相对误差(11%~18%)稍微大一点[5]。 因此, 由于植物样品基体的复杂性, 在利用XRF技术进行定量分析时, 需要采用与样品基体相匹配的标样来校正基体效应, 以及建立校准曲线, 从而获得较为准确的定量结果。
样品的整体分析仅代表样品的平均水平, 对于在局部微环境发生的生物化学反应还需从组织、 甚至细胞层面进行分析。 μ -XRF技术能够在微米尺度下实现元素的(半)定量分析, 通过在微观尺度上原位测定植物组织中元素的浓度和分布, 有助于认识植物对元素的吸收、 分配和富集机制。 目前, 表征植物样品中元素的微区分布研究, 大多是获得了某一区域内元素富集多、 少的相对关系, 而很少获得元素的浓度分布图, 这主要是由于植物样品基质的复杂性和异质性, 导致很难找到与样品的密度、 厚度和基体一致的标样[8]。
为获得植物样品中元素的定量分布信息, 在进行μ -XRF分析时, 常采用蒙特卡罗模拟[9]、 基本参数法[10]和Ray* Ray/Com或Ray/Com(瑞利与康普顿散射峰的比值)[11]进行基体校正。 如运用同步辐射共聚焦μ -XRF分析番茄根横截面中Fe的浓度分布时, 基于基本参数法和植物标样进行基体校正, 发现根表层中Fe浓度比根中部Fe浓度高2~3个数量级, 且根组织中Fe的浓度都低于10 μ g· g-1[12]。 由于10 μ g· g-1是大多数实验室微区分析仪器的检测极限, 故很难运用其他技术获得植物中元素的浓度分布, 尤其是进行快速的元素扫描(上述Fe浓度分布扫描时间约1 h)。 同时, 标准物质的参与对于评估定量分析所需的绝对检测灵敏度是必不可少的, 通过交叉叠加多层Mylar膜以及不同质量Co的方式, 在相似质量和厚度的状态下模拟了浮萍叶片的标样, 运用该标样进行的μ -XRF分析对活体浮萍叶中Co的检出限可达250 μ g· g-1[13]。
另外, 由于活体植物中水分对光子的衰减作用以及不同组织细胞中含水量的变化, XRF对活体植物的绝对准确定量仍然存在较大困难。 但有学者报道, 对于含水的生物样品, 连续发射X射线的散射作用所产生的强背景往往会降低其检测灵敏度, 通过构建质子诱导的准单色μ -XRF系统的方式能够增强单色X射线, 从而降低检出限, 其检出限甚至达到ng级[14]。 因此, 通过合适的校准方法和样品制备, XRF可以有效地提供活体植物中元素迁移与分布过程中的(半)定量信息。
从种子萌芽到植株生长过程中, 涉及到种子结构中元素的调动、 根系的元素吸收、 植物体内元素的运输、 以及细胞内元素的贮存和解毒等生物活动。 μ -XRF与XAS两种原位微区分析技术能够实时、 动态研究上述生物活动中元素的分布特征和分子形态, 从而揭示植物体内元素的生物行为和功能。
1.2.1 种子萌芽及幼芽生长过程中元素的动态变化
种子的萌芽过程会调动种子结构中储存的矿质元素, 同时也会影响毒性元素的分布和形态转化。 μ -XRF技术能够在种子萌芽过程中实时、 原位、 无损扫描元素的分布特征, 如μ -XRF分析发现, 萌芽期间细胞对矿质元素的调动能力呈现K> Ca> Zn> Mn> Fe的规律, K、 Ca被高度调动以促进新生根和叶原基的生长, 而Fe主要储存于液泡中, 其调动能力最低[15]。 在未发芽的小麦种子中, Zn和P均主要分布在糊粉层, 而在萌芽的种子中, 部分Zn从糊粉层迁移至种皮, 结合XNAES分析发现, 在种子萌芽过程中, 糊粉层中的锌-肌醇六磷酸络合物发生部分水解, 从而促进Zn从肌醇六磷酸结构中释放出来[16]。 种子的萌芽也会诱导毒性元素的迁移, 如菠菜和香菜种子从萌芽到形成新生根的过程中, Pb主要分布在胚根和新生根中, 且种皮、 胚乳、 新生根的底部中Pb主要以Pb5(PO4)3Cl或Pb3(PO4)2的形式存在, 而根尖中仅存在有机铅形态(乙酸铅和铅-富里酸络合物), 表明有机铅络合物是种子萌芽和幼芽生长过程中Pb的迁移和运输机制, 而铅-磷酸盐是种子对可溶性Pb的解毒和耐受机制[17]。 综上, μ -XRF和XANES技术是探索种子萌芽和幼芽生长过程中元素的微区分布和形态动态变化的有力手段。
1.2.2 植物根系对元素的吸收作用
植物根系对营养元素或毒性元素的吸收与根际环境条件密切相关, 如根际中元素的价态、 有机物和氧化还原条件等。 如植物对Se(Ⅵ )的吸收能力往往大于Se(Ⅳ ), 利用XANES技术分析其原因, 发现只有在Se(Ⅳ )暴露下, 24 h内豇豆根部大部分的Se转化为硒代蛋氨酸和硒-半胱氨酸, 表明植物体内形成迁移性较低的有机硒, 是导致植物易于吸收Se(Ⅵ )的主要原因[18]。 另外, 有机螯合剂也会影响元素的吸收过程, 如EDDS能够显著促进黑麦草根对Cu的吸收和运输, 其可使Cu从根到茎的迁移率增加6~9倍, 结合SRXRF和XANES技术分析发现, EDDS通过减少Cu在根尖分生组织和主侧根连接处的沉积, 同时驱动植物体内形成Cu-EDDS螯合物, 从而促进Cu向地上部运输[19]。 根系对元素的吸收还会受根际氧化还原条件的影响, 如与非淹水处理相比, 淹水条件下柳树根、 茎和叶中Cu的积累浓度分别降低了40.7%, 62.3%和48.2%, μ -XRF分析发现, 淹水条件下Fe和Mn在柳树根表皮外层呈圆环分布, 且表皮层中Cu的信号强度显著低于非淹水条件, 表明淹水条件下根表面形成的Fe/Mn膜抑制了柳树根对Cu的吸收[20]。 从上述研究实例, 可以发现不同价态的元素、 螯合剂和土壤淹水条件均通过促进元素在植物体内的转化或改变植物对元素的区隔机制, 来影响根系对元素的吸收。
1.2.3 植物中元素的运输和生物转化
植物中元素的运输主要包括元素从根表皮到中柱的迁移、 木质部的运输和韧皮部的运输三个过程。 首先, 根表皮到中柱的元素迁移过程是植物体内元素再分配的重要前提, 该过程需要元素依次穿过表皮、 皮层、 内皮层再到达中柱。 结合μ -XRF和XANES技术可表征元素从表皮到中柱的运输过程和形态转化, 如研究发现CuO NPs虽然难以穿过多数根内皮层的凯氏带进入中柱, 但仍可通过凯氏带结构被破坏的区域(主侧根连接处)进入中柱, 同时进入水稻的CuO NPs将部分转化为Cu(Ⅱ )与半胱氨酸、 柠檬酸和磷酸盐的络合物, 以及还原成Cu2O[21]。
随后, 元素通过水分的蒸腾作用经木质部从根部直接运输到地上部。 根、 茎、 叶柄横截面中元素的空间分布和木质部汁液中元素的浓度分析是揭示木质部中元素运输的重要途径, 如μ -XRF扫描发现Cu主要分布在杞柳根的中柱, 以及茎、 叶柄的维管组织, 同时相关性分析显示木质部汁液中Cu的浓度与茎中Cu的浓度显著正相关(r2=0.97, p< 0.01), 表明Cu主要通过木质部从杞柳根部运输至地上部[22]。 最后, 元素也可经韧皮部从老叶或成熟叶运输至新叶或新生根[23]。 如μ -XRF分析, 发现Cd在积累型东南景天老茎的韧皮部中也明显分布, 而在新茎的髓心和皮层中明显分布, 这有力地证明了韧皮部介导的Cd转运在Cd从老叶到新叶的再活化过程中起重要作用[24]。 因此, 植物的关键部位(中柱、 木质部和韧皮部)中元素的微区分布是揭示元素在植物体内迁移过程的重要依据。
为实现元素在木质部和韧皮部的长程运输中, 进入植物细胞的元素常发生形态转化, 尤其是转化为金属有机络合物[25]。 在大多数植物的韧皮部和木质部汁液中, 元素的浓度在μ mol· L-1 (几十~几百μ g· L-1)范围[26], 故在如此低浓度下, 准确鉴定韧皮部、 木质部中金属络合物的分子结构是阐明植物中金属元素运输过程的巨大挑战之一。 目前单配体的伏安滴定法和高效液相色谱-氢化物发生原子荧光光谱技术常用于分析木质部汁液中元素的形态, 但这两种方法可能存在配体吸附、 形成的络合物不稳定等问题, 从而影响形态分析的准确性[27, 28]。 然而, XAS技术能够解决上述问题, 并实现低浓度(μ g· L-1级)生物组织液的元素形态分析, 如利用同步辐射全反射X射线荧光扩展的XANES分析技术, 可以鉴定As浓度低至30 μ g· L-1的黄瓜木质部汁液中As的形态, 原位活体实验结果显示, 随着As暴露时间的延长(0~48 h), As的价态从As(Ⅲ )转化为As(Ⅴ )[29]。 因此, 依靠XANES技术分析木质部、 韧皮部中元素的形态是阐释植物中元素运输过程的重要途径。
1.2.4 植物中元素的贮存、 耐受和解毒机制
为降低游离金属离子对植物的毒害, 植物细胞可将重金属沉积在细胞壁和区隔在液泡。 细胞壁是元素进入细胞质的第一道屏障, 其组成成分对金属元素具有较强吸附能力, 如在pH 5.0~6.0范围内, 美洲商陆根细胞壁吸附的Mn高达5.4 mg· g-1, 同时XAS分析, 发现吸附到根细胞壁的Mn(II)通过内壳层配位与细胞壁的羧基结合[30]。 与之类似, 黑麦草在Pb暴露下, 根和叶细胞的Pb-L3 XANES形态分析, 发现大多数Pb(50%~65%)与细胞壁成分的多糖结合[31]。 拟南芥在Cd暴露下, 叶片表面毛状体细胞的Cd-L3 XANES形态分析, 发现75% Cd与细胞壁中的O/N配体结合, 仅25% Cd与细胞壁中含S的配体结合[32]。 为阐明海州香薷根细胞的细胞壁中Cu的结合配体, 有学者利用改性的细胞壁吸附Cu2+, 发现细胞壁的羟基、 羧基和氨基是Cu2+的主要结合位点[33]。 因此, 细胞壁中的羟基、 羧基和氨基等带负电的配位基团, 通过与金属离子发生离子交换、 吸附、 配位络合和沉淀等作用, 使进入植物细胞的金属元素沉积于细胞壁[34]。
当植物细胞壁上的结合位点与金属离子的结合量达到饱和后, 多余的金属离子将穿过质膜进入细胞内部。 细胞内液泡的区隔化是植物细胞对金属元素的另一重要解毒机制[35]。 如XANES分析发现, 当秋茄根细胞壁对Cd的固定趋于饱和后, 细胞内大多数的Cd2+被运输至液泡, 并与柠檬酸和草酸络合[36]; 在Pb胁迫下, 绿藻细胞中约10% Pb与GSH结合, 形成Pb(GSH)3络合物[37]; 拟南芥分别暴露在As(Ⅲ )和As(Ⅴ )环境中, 在其叶细胞中鉴定出32.0%和18.9% As(Ⅲ )-GSH[38]; 紫花苜蓿在Hg暴露下, 其根细胞中Hg以43% Hg-PC, 34% Hg-Cys和23%甲基汞形式存在[39], 表明液泡中的谷胱甘肽(GSH)、 半胱氨酸(Cys)、 有机酸和植物螯合肽(PC)等通过与金属元素形成低毒性或无毒的有机络合物, 以实现元素的细胞解毒。
X射线光谱技术也广泛应用于微生物、 动物和人体组织中元素的分布和形态研究, 结合μ -XRF和XANES技术可研究微生物作用下元素的氧化还原、 生物矿化和沉淀固定, 探索动物与元素的交互作用及其对重金属环境的适应机制, 以及揭示元素在人体组织中的代谢行为等。
微生物普遍存在于环境中, 在元素的生物地球化学循环过程中扮演着重要角色, 其可通过对重金属的溶解、 吸附、 形态转化等途径影响元素的循环过程。 结合XRF和XAS技术, 可探究微生物作用下元素的地球化学循环过程, 并揭示微生物对重金属的解毒机制, 从而为重金属污染的生物修复提供理论依据。
2.1.1 微生物作用元素的迁移过程
砷的氧化还原机制、 砷在固相和液相之间的循环过程、 以及砷从沉积物中释放进入水体的机理等一直是高砷沉积物和地下水研究的重要内容。 通常, As(Ⅲ )比As(Ⅴ )的毒性更大, 无机砷化合物的毒性强于有机砷化合物[40]。 XANES分析发现在还原条件下, 溶液中的砷主要以As(Ⅲ )形式存在, 固相中吸附的砷也有74%~85%以As(Ⅲ )的形式存在, 表明具有较高的生态风险; 当环境中加入氧气后, 在原生微生物的作用下, 溶液中As(Ⅲ )的含量降低, 但As(Ⅴ )的含量并未增加, 表明氧化条件可降低砷的溶解性, 使其被还原为As(Ⅴ )并吸附于固相, 从而降低As的生态风险[41]。 另有研究利用XANES技术研究含铁矿物中As的吸附-解吸附行为, 发现缺氧的稻田土壤中分离的地衣杆菌Geobacter sp.OR-1可促进吸附于水铁矿中砷的异化还原与释放, Geobacter sp.OR-1作用可将As(Ⅴ )还原为As(Ⅲ ), 也可将Fe(Ⅲ )还原为Fe(Ⅱ ), 导致原本吸附于水铁矿表面的砷释放进入稻田环境中, 从而增加了As进入食物链的生态风险[42]。 因此, XANES技术可用于研究微生物作用下As的迁移和转化过程, 并揭示矿物表面As的吸附与释放行为。
2.1.2 微生物作用元素的形态转化
微生物促进毒性元素发生氧化还原和形成有机络合物是微生物适应环境并存活的重要途径。 如利用XAS技术研究非活性的枯草杆菌和假单胞菌对Au的转化, 发现由于Au(Ⅲ )与微生物细胞表面的链酶发生电子转移, 导致90%的Au(Ⅲ )被还原为Au(Ⅰ ), 且Au(Ⅰ )与巯基结合的比例随时间的延长而增加, 表明巯基对Au的还原起着重要作用[43]。 与之类似, 利用XRF和XANES技术研究贪铜菌细胞内Au的分布和形态特征, 发现Au主要以Au纳米颗粒(NPs)的形式聚集在细胞中心[44]。 除此之外, 微生物的胞外分泌物也在促进重金属形态转化和降低毒性胁迫中发挥重要作用, 如XANES分析发现, 当草酸青霉菌暴露于Cr(Ⅵ )溶液中, 细胞分泌的含巯基化合物(半胱氨酸)和草酸增加, 导致Cr(Ⅵ )被还原生成草酸铬, 同时与半胱氨酸结合形成Cr(Ⅲ )-Cys[45]。 外源Ca2+也能促进草酸青霉菌产生胞外分泌物, 并在细胞表面形成大量的草酸钙, 通过有效吸附Cr, 以防止Cr进入细胞造成毒害; 外源
2.1.3 微生物促进重金属的修复
微生物通过促进重金属的生物矿化将重金属沉淀, 从而降低重金属的生物有效性, 这是微生物修复重金属污染的重要途径之一。 生物矿化主要包括磷酸盐矿化、 碳酸盐矿化和硫化物矿化等。 Stylo等[47]利用XAS探究了希瓦氏菌Shewanella oneidensis MR-1对土壤中U(Ⅵ )的生物矿化过程, 发现希瓦氏菌可将U(Ⅵ )转化为溶解性较差的UO2矿物, 环境中加入S, Si和P元素可促进胞外聚合物(EPS)的形成, 由于EPS可提高细胞对U(Ⅵ )的吸附及促进成核, 同时这些EPS中的细胞色素可促进U(Ⅵ )的还原, 使被还原矿化形成的UO2比例增加55%~95%, 从而有利于U(Ⅵ )的生物修复。 另有学者结合XRD和EXAFS技术研究了真菌作用铀矿物的氧化物溶解和生物矿化过程, 发现暴露于UO3和U3O8的真菌通过分泌大量的草酸, 促进U的溶解和(UO2)2+络合物的形成, 同时可溶的(UO2)2+进一步与磷结合, 形成UO2HPO4或[UO2(HPO4)2]2-, 并最终矿化形成结晶度较好的磷酸铀酰矿物[48]。 与之类似, 利用XRS技术探究草酸青霉SL2(Penicillium oxalicum SL2)对Pb的修复机制, 发现Pb2+暴露后, 细胞分泌的草酸、 柠檬酸、 磷酸氢根、 GSH均有明显的增加, 菌丝体胞内形成了纳米级和微米级的含Pb次生矿物, 主要为草酸铅、 柠檬酸铅、 磷酸氢铅及Pb-GSH(铅-谷胱甘肽)络合物, 由此判断GSH的合成、 有机酸的分泌和有机磷的水解作用促进了Pb的形态转化和生物矿化[49]。
底栖动物(蚯蚓等)在生态系统的物质循环中发挥着巨大作用, 其不仅能够耐受并富集环境中的(类)重金属元素, 还可以通过自身活动改善土壤环境和性质, 从而对植物生长和土壤微生物菌群等产生影响。 μ -XRF和XAS技术在原位测定元素分布和形态方面具有独特优势, 能够为底栖动物与元素作用机制研究提供关键信息。
2.2.1 蚯蚓与土壤生态系统
底栖动物与土壤生态系统的交互作用能够影响土壤中(类)重金属的迁移和环境行为。 如研究蚯蚓和豆科植物对土壤硒行为的交互作用(图2), 发现蚯蚓作用下土壤Se向植物的迁移率增加4%, 进一步分析Se的XANES形态, 发现在硒酸盐环境下, 蚯蚓的存在使植物中有机硒形态(如SeMet和CysSeSeCys)的百分比大大增加(高达34%), 而单质Se的比例明显降低, 表明蚯蚓明显增加了植物对Se的吸收和转移, 从而提高植物地上部的Se积累[50]。 在污染的土壤中引入蚯蚓可改变土壤中元素的形态、 提高元素的可迁移性和生物可利用度, 比较三种类型的蚯蚓(Eisenia veneta, Lumbricus terrestris和Allolobophora chlorotica)对污染土壤中Cu, Pb, Zn和As元素的迁移性和可利用性的影响, 并在土壤表面种植黑麦草来评估植物对重金属的吸收, 发现L.terrestris和E.Veneta不仅能够增加Cu和Zn的迁移率, 还能增加有效性更高的金属形态(金属的自由离子)的比例; 另外, 相对于其他两种类型的蚯蚓, L. terrestris能够产生更多的土壤排泄物, 并促进黑麦草对重金属的吸收[51]。
 | 图2 蚯蚓与土壤生态系统中Se的迁移转化[50]Fig.2 Translocation and transformation of Se in earthworm and soil ecosystem[50] |
2.2.2 蚯蚓中金属元素的生物过程和行为
蚯蚓常摄食和分解凋落物, 以实现生态环境中养分和毒性元素的循环。 为阐明蚯蚓从土壤中吸收、 转运和富集重金属的生物过程, 结合μ -XRF和XAS技术研究土壤和蚯蚓中Zn和Pb的分布特征和形态转化, 发现在蚯蚓前、 后消化道均有Zn的分布, 而Pb主要分布在后消化道的黄色体细胞中, XANES分析显示土壤和蚯蚓中Zn的形态相似, Zn主要与氧配体结合, 其结构与Zn3(PO4)2相似, 其次与S配体结合[52, 53]; 然而, 土壤和蚯蚓中Pb的形态存在差异, 土壤Pb呈单壳层配位结构(Pb-S), 而蚯蚓的黄色体细胞中的Pb呈双壳层配位结构(Pb-O与Pb-S), 表明蚯蚓的黄色体细胞在Pb的解毒和富集中起重要作用, 而且蚯蚓对营养元素Zn和毒性元素Pb的吸收、 代谢过程存在显著差异[52]。 与之类似, 蚯蚓暴露于含Ag NPs的土壤环境中, μ -XRF分析发现Ag主要分布于消化道表面的氯生组织、 肾小管以及刚毛附近[54], 同时XANES分析发现蚯蚓体内的Ag可能与富含硫醇的蛋白质结合[55]。 上述研究表明, 元素的分布和形态研究是揭示动物中元素的生物过程、 解毒和富集机制的重要途径。
2.2.3 其他动物组织结构中元素的分布和形态
为探索蚤状钩虾对高As环境的适应和生存机制, 结合μ -XRF和μ -XANES技术研究蚤状钩虾从叶片摄食、 消化和贮存As的过程, 发现As主要沿蚤状钩虾的肠道系统分布, 并在作为食物来源的叶片中, 鉴定出68%砷酸盐和29%一甲基砷酸盐, 摄入肠道系统后, 叶片中的As被转化为46%~56%二甲基砷酸盐和23%毒性更高的亚砷酸盐, 而肠道外的其他组织中, As主要以甲基砷的形式存在, 其次是As(Ⅲ )-S络合物(10%~21%), 表明食物中的主要As形态(砷酸盐)经蚤状钩虾消化, 并在肠道发生甲基化和还原, 随后进入其他组织进一步甲基化或与S结合[56]。
含锶药物对骨质疏松症患者具有治疗作用, 为探索含锶药物如何在骨骼中发挥作用, 有学者利用2D μ -XRF和3D同步辐射μ -XRF技术扫描了雷尼酸锶和柠檬酸锶处理的大鼠肱骨中锶元素的分布, 发现两种锶盐处理下, 锶具有相同的空间分布, 其大量沉积于肱骨骺板下方的骨小梁区域, 且肱骨中高钙分布区域与低锶分布区域相对应, 同时两种锶盐处理下, 锶层厚度(1.364~1.382 mm)无显著差异(p=0.920 1), 表明锶盐作用下新形成的骨骼能够通过钙原子的取代或表面交换将锶掺入羟基磷灰石晶体中, 从而增加骨骼的强度和降低骨质疏松性骨折的风险[57]。
研究人体组织中重金属的浓度、 分布和形态特征, 有助于揭示重金属对人体健康的影响和人体内重金属的代谢过程与作用机制, 人发和指甲作为人体的附属组织, 可直接采集离体组织用于元素的分析测试; 而骨骼常位于真皮组织层下, 其元素的分析测试还依赖于原位活体的分析方法, 如活体XRF原位分析技术。
2.3.1 人发和指甲
人发和指甲中元素的分布和形态特征能够反映一段时间内元素在人体内的吸收和代谢情况。 例如, μ -XRF技术扫描矿区采集人发样品中元素的分布, 发现Pb和As主要沿头发中轴分布, 且从发根至发梢逐渐增多, XANES分析元素的形态, 结果显示人发中的Pb由54.7% Pb3(PO4)2和36.8% Pb-GSH和8.4% PbS组成[58], As由73% As(Ⅲ )-S和27% As(Ⅴ )
2.3.2 人体骨骼
活体XRF分析技术通过直接测定人骨中毒性元素的含量和获得骨细胞中毒性元素的原位信息, 以探究毒性元素对人体健康的影响及其体内代谢过程与作用机理。 如有学者采用109Cd激发Pb的K系谱线, 原位活体分析了人体胫骨和根骨中Pb的含量, 发现普通人群的骨铅浓度在0.4~22.7 μ g· (g 骨矿物质)-1, 而铅锌矿区附近居民骨铅含量较高, 其Pb浓度高达73.9 μ g· (g 骨矿物质)-1, 表明采矿活动潜在威胁着周边居民的人体健康。 摄入人体的毒性元素也会进入骨细胞中, 影响骨细胞的存活和功能。 如通过结合μ -XRF和XANES技术研究Cr进入骨细胞的过程、 细胞分布和形态特征, 发现Cr3+暴露下, 进入成骨细胞的Cr3+聚集在细胞核及细胞核周围, 并与羟基和磷酸根结合, 形成Cr(OH)3和CrPO4; 而Cr6+暴露下, 进入成骨细胞的Cr6+分散于细胞内, 并完全转化成Cr3+, 表明Cr3+和Cr6+的骨细胞作用机制存在显著性差异, 且细胞内Cr6+还原成Cr3+、 以及氢氧化铬和磷酸铬的沉积是成骨细胞对Cr的主要代谢和解毒机制[61]。 因此, XRF和XANES技术是原位监测和分析毒性元素对人体骨骼影响和相关机理的有效手段, 其可为诊断重金属在人体中引起的生物致病机制提供直观数据。
海洋、 土壤沉积物和大气颗粒物等是环境中营养元素和毒性元素的重要载体。 获得各载体样品中元素的组成、 微区分布、 形态特征以及矿物结构信息是揭示环境中元素的迁移、 转化和累积等环境行为的关键。 近年来, X射线光谱技术常用于监测和评估重金属的环境风险、 研究环境微粒物对重金属的载运机制和解析重金属的环境归趋、 演化等。
大气环境中, 能量色散X射线光谱(EDXRF)和全反射X射线荧光光谱(TXRF)常用于大气重金属污染的植物监测和重金属污染源的识别。 汽车尾气和大气颗粒物是大气环境中的两大主要污染物, 其元素组成、 浓度和形态特征是影响大气中重金属污染程度的重要因素。 μ -XRF和XANES技术常用于衡量元素在大气环境中的迁移潜力、 解析重金属的污染来源以及识别大气颗粒物中矿物的转化。
3.1.1 大气中重金属污染的监测
植物监测为大气环境的污染评估提供了一条便捷而有效的途径, 苔藓、 植物叶片和树皮已被广泛应用于大气中重金属的污染监测。 如利用EDXRF技术检测3年间苔藓样品中元素的浓度变化, 发现3年间Br, Ca, Fe, Zn, Rb, Sr和Pb的浓度无显著性差异, 表明已知的污染源无显著变化和/或没有新的污染源[62]。 植物叶片常充当气溶胶和气体颗粒物的沉积池, 利用TXRF技术直接测定6种树木叶片中元素的浓度, 发现榆树是最适合用作生物指示剂的树种, 马可尼公园是大气污染风险最高的公园, 同时利用多元统计分析树叶中沉积重金属可能的来源, 发现车辆排放物是Cu, Fe和Pb元素的主要污染源[63]。 另外, 树皮也常用于监测大气污染和识别大气的污染源, 在EDXRF技术测定树皮中元素浓度的基础上, 应用主成分分析识别元素的污染源, 发现树皮中Al, Ba, Cu, Fe, Mn和Zn主要来源于汽车刹车、 轮胎的磨损以及道路扬尘的再悬浮[64]。 上述研究实例表明, XRF技术在大气污染物的生物监测和重金属污染源的识别中发挥了重要作用。
3.1.2 汽车尾气中元素的环境风险
汽车尾气烟尘常携带重金属、 硫化物和氯化物等, 这些携带物可对汽车行驶道路周围的土壤/粉尘、 植被和空气产生污染。 为评估汽车尾气烟尘中重金属对大气环境的危害, 利用EDXRF测定了汽车尾气烟尘中元素的浓度, 同时以Ca为参考元素计算样品中元素的富集因子(EFcrust), 发现Cr, Mn, Ni, Cu, Zn, Br和Pb在汽车尾气烟尘中高度富集(EFcrust> 10), 尤其是重金属Cr, Pb和Zn的污染最为严重, 三者的EFcrust分别高达696, 72.4和183[65]。 元素的形态分析可进一步衡量元素在大气环境中的迁移能力和生态毒害。 如利用XNAES技术鉴定车辆废气颗粒物中Pb和Zn的形态, 发现Zn主要以ZnCl2的形式存在, Pb主要由3种化合物组成, 包括52% Pb3(PO4)2, 24% PbSO4和23% PbCO3, 由于ZnCl2易溶于水, 而Pb3(PO4)2和PbSO4在环境中较稳定, 故车辆废气颗粒物中Zn的迁移性较高, 而Pb的迁移能力相对较低[66]。 上述研究表明, 结合EDXRF和XANES技术, 可从元素总量和形态层面准确评估汽车尾气烟尘中元素对生态环境产生的危害程度。
3.1.3 大气颗粒物的元素组成及来源解析
大气颗粒物是重要的空气污染物之一, 其环境毒害程度取决于颗粒物的尺寸、 元素组成、 颗粒物的粒径分布和元素形态等。 如利用μ -XRF技术探究不同粒径的大气颗粒物中元素的分布特征, 发现Ca和Ti主要分布在> 2 μ m粗粒径颗粒中, 而V, Cr, Mn, Ni, Zn, Cu, Pb, Cl和S主要分布在0.1~1.0 μ m的颗粒物中, 且这些元素的富集程度随粒径的增大而减小[67]。 另有研究通过对比冶金工厂附近PM10和PM2.5中As的浓度和形态, 探究了不同粒径的大气颗粒物中As的潜在毒性, EDXRF测试发现PM10中As浓度的平均值(30 ng· m-3)比欧洲目标值高4倍, 而PM2.5中As的浓度是PM10中As浓度的一半, 但两种颗粒物中As的潜在毒性不完全取决于As浓度的高低, 还与As的形态密切相关, XANES分析发现, PM10中As以As(Ⅴ )为主, 而PM2.5中As以毒性更高的As(Ⅲ )为主, 故相比大粒径的颗粒物(PM10), 小粒径的颗粒物(PM2.5)具有更高的As毒性[68]。 与之类似, 利用XANES技术表征粗(> 2.1 μ m)、 细(≤ 2.1 μ m)大气颗粒物中Pb的形态, 通过与道路灰尘、 垃圾焚烧飞灰和燃油飞灰中Pb形态进行对比, 发现粗颗粒中的Pb主要来源于道路灰尘, 而细颗粒中的Pb主要来源于城市生活垃圾焚烧和重油燃烧产生的飞灰[69]。 综上, μ -XRF和XANES技术是评估大气颗粒物中毒性元素的危害潜力和污染来源的有力工具。
X射线光谱技术在海洋微塑料、 海洋沉积物和海洋动植物的元素分析中应用广泛, 利用XRF技术定量分析海洋环境样品中元素的浓度, 可准确评估海洋环境中重金属的生态风险, 同时结合μ -XRF和XANES技术探索铁锰结核、 锰-硫化物和富稀土底泥等沉积物中元素的分布和形态, 能够揭示海底沉积矿物的形成机制及其吸附重金属、 稀土元素的过程; 表征海洋典型动植物中元素的分布和形态特征, 不仅有助于认识海洋动植物细胞中金属的富集和解毒机制, 还可为古海洋环境演化研究提供帮助。
3.2.1 海洋微塑料中元素的生态风险
微塑料是海洋环境中最重要的污染物之一, 其不仅会通过缠结和吞咽作用直接影响海洋生物的生存, 还会作为运输和生物蓄积有机、 无机化学物质的媒介, 间接威胁海洋生物的生存。 快速获取微塑料中元素的定量信息是准确评估微塑料潜在生态风险的重要前提。 近年来, 配置低密度模式和小光斑设备的便携式X射线荧光(FP-XRF)光谱仪常用于测定微塑料中元素的浓度, 如有学者利用FP-XRF分析微塑料中元素的浓度, 发现海滩微塑料中Cd, Cr和Pb的浓度分别高达4 310, 1 330和6 130 μ g· g-1, 其分别超过《欧盟有害物质限制指令》所规定限制值的43.1, 1.3和6.1倍[70], 进一步评估微塑料中元素的生物可给性, 发现模拟海鸟胃液提取液中Cd和Pb浓度分别高达50和8 μ g· g-1, 其分别比海鸟可食用的安全阈值高出50和4倍[71]。 除了Cd, Cr和Pb污染外, Br和Sb也是微塑料中常见的污染元素, 其主要源于含Br和Sb阻燃剂的使用, FP-XRF测定微塑料中Br和Sb的浓度, 显示两者的平均浓度高达1 410和621 μ g· g-1, 且模拟海鸟胃液提取液中Br和Sb浓度分别高达14和1.4 μ g· g-1, 表明微塑料所运载的毒性元素潜在威胁着海洋生物的生存[72]。
3.2.2 海洋沉积物中元素分布与物源识别
海洋铁锰结核中富铁和富锰层呈现出洋葱状交替分布模式, 海底水环境季节性循环是产生该分层模式的主要因素[73]。 在春季, 海底水中氧气降低, 进入结核的Mn通量超过Fe通量, 此时的亚氧条件更有利于Mn4+还原成Mn2+; 在夏季, 底部海水中缺氧的增加, 将导致进入结核的Fe通量高于Mn通量, 促进富Fe层的沉积[74]。 这些富铁和富锰层可吸附海洋中的微量元素, μ -XRF和XAS是研究铁锰结核中微量元素吸附和分配的重要手段, μ -XRF扫描发现Zn主要吸附在富Mn层, 而As主要吸附在富Fe层, EXAFS分析表明海底铁锰结核中的锰相主要是结晶性较差的水钠锰矿, 其与土壤中报道的锰氧化物相似, Zn易于吸附到水钠锰矿上, 并形成四面体的配位结构, 这主要是由于四面体结构比八面体结构更好地补偿由Mn3+取代Mn4+引起的层电荷不足, 而As主要以As(Ⅲ )吸附在水铁矿上[75]。 因此, 探究海底铁锰结核中元素的分布和形态, 可揭示铁锰结核的生长模式和形成环境。
探索海洋沉积物中Mn的富集机制是揭示锰-硫化物形成的重要途径, 结合Micro-XRF和SEM-EDS技术, 发现块体叠层中Mn的分布规律与Fe和S的分布一致, 表明硫化物结合的Mn与草莓状黄铁矿聚集体有关[76]。 为进一步探究硫化物结合的Mn是否来源于Mn对内生铁-硫化物矿物的同构替代, 利用EXAFS技术表征了沉积物中Mn的配位结构, 发现Mn— S键长(2.3 Å )大于黄铁矿中Fe— S键长(2.2 Å ), 但与四方硫铁矿中Fe— S的键长(2.26 Å )相近, 同时鉴定出样品中Mn与S的配位结构(四面体)与黄铁矿中Mn— S的配位结构(八面体)不同, 而与四方硫铁矿中Fe— S的配位结构(四面体)相同, 表明硫化物结合的Mn未进入黄铁矿结构中, 而是同构替代了四方硫铁矿中的Fe[76]。 另有研究发现大洋沉积物中钇与铁的空间分布一致, 由于铁信号主要来源于热液活动形成的氢氧化铁颗粒, 故沉积物底泥中的钇可能来源于大洋中脊的热液活动[77]。 因此, 元素的分布、 价态和原子的配位结构有助于阐释海底沉积矿物的形成机制和潜在来源。
3.2.3 海洋典型生物对海洋环境的生物指示作用
有孔虫是一种海洋生物指示剂, 其碳酸钙壳体中的Ca, Mg和Zn等微量元素能够指示水环境的温度、 海洋演化、 全球环境与气候变化等。 有孔虫壳体方解石的Mg/Ca比值常用作指示海洋温度, 较高的温度有利于镁取代钙吸热, 但有孔虫中Mg的掺入可能更为复杂, XANES分析发现有孔虫方解石的矿物结构与白云石((CaMg)(CO3)2)相似, 且Ca和Mg在晶格中占据等效八面体位置, 表明Mg可以替代有孔虫方解石中的Ca[78]。 有孔虫壳体方解石的Zn/Ca比值常用于认识海洋水化学和古环流, 结合μ -XRF和XANES技术探索有孔虫壳体中Zn的掺入机制, 观察到壳体中Zn和Ca的分布相似, 且50%的Zn以钙替代物或以四配位氧吸附离子的形式存在于方解石中, 部分Zn以闪锌矿(形成于早期成岩过程)和磷酸锌的形式存在, 还有少量的Zn以水锌矿的形式存在, 由于水锌矿来源于有孔虫的细胞过程, 故有孔虫壳体中Zn的掺入也与其细胞过程相关[79]。 综上, 有孔虫壳中元素的分布和化学形态可揭示微量元素从海水环境到有孔虫钙化位点的迁移和掺入过程。
珊瑚中微量元素S, P, Ca和Mg的分布和形态能够指示珍贵珊瑚的生长特征和栖息地的环境。 据报道, 珊瑚骨骼中的Mg可指示海水温度, 其含量随海水温度的升高而增加, 海水温度每升高1 ℃, 珊瑚骨骼中Mg/Ca比值将增加0.004~0.006[80]。 珊瑚的生长会形成明亮(低有机质区)和黑暗(高有机质区)交替的生长环带[81], μ -XRF分析显示, 红珊瑚骨骼中S, P和Mg沿生长环的明暗带分布, S和P主要分布在富有机质的暗带中, 而Mg则分布在低有机质的亮带中[82]。 进一步的XANES形态分析, 发现CaSO4是珊瑚骨架和共胶层中S的主要形态, 即S能够以硫酸盐的形式取代碳酸根离子进入方解石骨架中, 同时在XANES光谱图的2 480.1 eV处观察到一个较低的能量峰, 表明珊瑚骨架中也存在硫酸氢盐或硫酸软骨素, 由于硫酸软骨素是一种粘多糖, 其常以蛋白聚糖的形式附着在动物组织的蛋白质上, 故珊瑚中的S能够以无机和有机硫形式共存[83]。 上述研究表明, 元素分布和形态分析有助于更准确地认识珊瑚中S元素的环境行为, 并为探索S元素在生物矿化中的作用提供重要信息。
土壤和沉积物体系中, μ -XRF, XANES和μ -XRD技术被广泛应用于评估元素的生物有效性、 阐释有机质改变元素迁移潜力的作用机制、 研究金属纳米颗粒物的迁移和形态转化, 以及探索磁性粒子对重金属的载运行为。
3.3.1 土壤中元素的生物有效性
元素的形态鉴定是准确评估土壤中元素的生物有效性的重要方式。 虽然化学提取法是表征土壤中元素生物有效性的一种常用手段, 其可提供土壤中元素形态分布的半定量信息, 但其无法提供元素的配位结构信息[84]。 结合XANES技术和化学提取手段表征矿区周围土壤中As的形态, 发现土壤As主要以砷酸盐矿物的形式存在, 其结构与水砷铁石相似, 同时还存在部分砷菱铅矾; 顺序提取也表明土壤As主要与结晶态的Fe氧化物结合, 结晶态的Fe氧化物的溶解度比非晶态的Fe氧化物低5倍[85], 故两种方法一致表明, 该矿区土壤中As的迁移潜力较低[86]。 相比矿区土壤, 农业土壤中元素的生物有效性较高, 其根本原因是两类土壤中元素形态的差异, XAS分析发现在矿区土壤中, Cu(Ⅱ )主要通过内壳层络合作用吸附到Fe(Ⅲ )氧化物上[87]; 而在污染的农业土壤中, Cu(Ⅱ )主要与土壤有机质(SOM)络合, 并形成Cu— N或Cu— O的双齿络合物, 这主要是由于农业土壤中植物的根系作用, 导致农业土壤中形成的SOM较多(2.2%~75%)[88], 而矿区土壤中SOM较少(~0.55%)[87]。 因此, 虽然矿区土壤中Cu的浓度远高于农业土壤, 但矿区土壤中Cu的生物有效性往往低于农业土壤, 故与元素的总量相比, 元素的形态更能直观地反映出土壤中元素的迁移性和生物有效性。
3.3.2 土壤有机质对元素迁移能力的影响
有机质提供的有机配体能够与土壤中的金属元素发生络合和吸附作用, 从而改变土壤中元素的迁移能力和生物有效性。 有研究者结合顺序提取和XANES技术研究了茶多酚(TP)对土壤中Pb形态的转化和Pb迁移能力的影响, 发现相比无TP处理组, 添加TP使茶叶土壤中可交换态Pb、 吸附态Pb和可还原态Pb的比例分别降低了1.5%, 9.1%和5.6%, 而有机结合态铅(Pb-TP和铅-富里酸)的比例增加了29.7%, 同时残渣态铅(Pb5(PO4)3Cl)的比例也增加了6.6%, 表明TP的添加显著改变了土壤中Pb的形态, 并降低了土壤中Pb的可迁移性[89]。 XAS分析还发现在低Pb浓度下, 1个Pb2+易与2个来自不同腐殖酸(HA)分子的羧基(COO— )配位, 并形成COO— Pb(Ⅱ )— COO— 桥型结构的二齿或多齿络合物, 通过促进腐殖酸分子之间的聚合, 来增加铅-腐殖酸络合物的稳定性, 从而降低土壤中Pb的可移动性[90]。 与之相反, 有报道称添加HA能够将污染土壤中Pb的生物有效性提高10%~30%, XANES分析其机理, 发现HA通过将稳定的磷氯铅矿(22%)和白铅矿(19%)转化为可迁移的Pb-HA络合物, 以提高土壤中Pb的生物有效性[91]。 因此, 土壤有机质与重金属的配位结构具有复杂性和不确定性, 其可能降低或升高土壤中重金属的可迁移性和生物有效性。
3.3.3 土壤中金属NPs的迁移和转化
土壤中NPs因其组成和纳米尺度, 导致其生态毒性往往比溶解态金属离子的毒性更大, 故NPs具有较高的迁移潜力和生物链传递风险。 如结合μ -XRF和μ -XANES技术, 分析发现ZnO NPs从土壤迁移到大豆后, 主要是以具有Zn— O配位结构的锌-柠檬酸盐存在于茎的韧皮部; 而CeO2 NPs从土壤迁移到大豆后, 79%的Ce仍以纳米形式存在于茎结节的表皮中, 同时还有部分Ce(Ⅳ )还原成Ce(Ⅲ )[92]。 类似的, 利用μ -XRF和μ -XANES技术研究农业土壤中Ag-NPs的命运, 发现土壤中的Ag-NPs易发生硫化, 形成极难溶解的Ag2S, 当种植小麦4周后, 10% Ag2S转化成银-硫醇化合物和无定型Ag2S, 表明小麦的根际作用促进土壤中Ag2S的形态转化和部分溶解[93]。 湿地土壤中NPs的迁移转化行为研究, 发现添加1 000 mg· kg-1 CuO NPs可使CaCl2提取水稻土壤中Cu的浓度提高374倍, 同时土壤中Cu的分子形态受水稻生长期的影响: 幼苗期, 土壤中71.5% CuO NPs转化成铜-腐殖酸结合态; 成熟期, 土壤中的CuO NPs完全转化成铜-腐殖酸、 Cu2S和Cu2O[94]。 因此, 在土壤-植物系统中, 植物的根系作用通过促进土壤中NPs的形态转化, 来提高土壤中金属NPs的可迁移性和生物有效性。
3.3.4 土壤中重金属磁性载体的环境意义
土壤中磁性粒子(TMPs)是重金属污染物的重要载体, 在微米尺度上认识TMPs中Fe的矿物相和重金属的空间分布, 有助于阐释污染土壤中TMPs的磁性特征及其与重金属的作用关系。 如μ -XRF技术扫描钢铁工业区污染土壤中微米级TMPs中元素的分布, 发现Co, Cr, Mn, Ni和Pb共同分布在8~96 μ m大小的富Fe区域, 结合XANES分析, 表明磁铁矿和铁合金是TMPs中主要的磁性来源和重金属载体[95]。 单个球形TMPs(直径30 μ m)的μ -XRF分析, 还发现TMPs中富含Fe, Pb, Cd, Zn和Cu, 进一步的μ -XRD分析表明这些重金属离子通过取代磁铁矿或赤铁矿晶格中的Fe2+和/或Fe3+而掺入到铁氧体结构中, 且铁氧体的磁性越高, 其结合重金属的能力越强[96]。 为确定土壤中的重金属来自何种磁性相, 结合μ -XRF和μ -XANES分析, 发现钢渣和煤灰中的铁合金、 磁黄铁矿和TMPs是污染土壤中的主要磁性相; Co, Cr, Cu和Mn主要来自铁合金, 而Ti、 Zn和As来自煤灰的铁合金和TMPs[97]。 因此, TMPs能够吸附土壤中的重金属, 可作为重金属来源的示踪剂, 揭示土壤中重金属污染物的来源。
纵观近年来国内外学者对生物地球化学与生态环境中元素的迁移规律、 环境行为和生物功能的研究, 可以发现(1)通过合适的校准方法和样品制备, XRF可有效地提供活体生物样品中元素迁移与分布过程中的定量数据; (2)μ -XRF和XAS技术是能够表征生态环境样品中亚微米尺度的元素分布和原子尺度的元素形态, 其原位分析特征在克服因样品制备导致的伪象中独具优势; (3)μ -XRF和XAS技术的联合应用可深入认识生物与元素的相互作用, 尤其是生物对元素的吸收、 转运、 贮存和解毒机制, 同时也能够揭示典型环境样品(如海洋铁锰结核、 土壤中磁性粒子和大气颗粒物)中元素的来源、 演化和归趋等环境行为; (4)结合全反射X射线荧光的X射线吸收近边结构谱技术能够成功鉴定低浓度(μ g· L-1级)的生物组织液中重金属元素的形态。
虽然μ -XRF和XAS技术已广泛用于生物与生态环境领域, 但由于生态环境样品基质的复杂性和多样性, 其仍存在一些技术难点与挑战:
(1)μ -XRF扫描样品中不同元素的分布时, 存在X射线荧光的自吸收效应, 尤其是对于轻元素。 如比较一个含水的植物样品(约0.1 mm厚)中Zn和P的分布, 对Zn而言, 基本能够检测到整个样品深度的Zn荧光信号(0.1 mm深度处吸收了约7%的荧光信号); 而对P而言, 在样品的较深处基本检测不到P的荧光信号(0.1 mm深度处吸收了≥ 99%的荧光信号) [98]。
(2)XANES难以准确定量样品中低丰度(5%~10%)的元素形态, 同时其数据的线性拟合高度依赖于标样, 而生物和环境样品的基质复杂, 较难确定样品中元素的配位基团, 以致于难以准确地选定参与拟合的标样, 同时目前用于拟合的标样有限, 尤其是缺少含有特殊基团的生物标样。 因此, 结合其他的元素形态分析技术(如非变性的色谱技术)和开发更多的标样是实现准确的XAS形态分析的发展方向。
(3)目前, 快速、 短暂的氧化还原反应(活细胞中元素的氧化还原循环)还难以用XAS来研究, 但X射线自由电子激光技术(XFEL)的发展可为研究这种快速的化学变化开辟新的研究领域, 其飞秒脉冲的特征非常适用于生物活性金属中心的时间分辨研究[99]。 虽然XFEL技术的样品破坏性限制了X射线信号的成像, 但其可弥补同步辐射X射线光源无法实现的瞬变氧化还原反应中元素的形态鉴定[100]。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|