
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
离子对反相液相色谱-原子荧光光谱法测定畜禽肉中5种硒形态含量
[魏益华1  , 黄青青
, 黄青青2 , 张金艳1, * , 邱素艳1, 3 , 涂田华1 , 袁林峰1 , 戴廷灿1 , 张标金1 , 李伟红1 , 严寒1 ]
, 黄青青, 邱素艳|
|


作者简介: 魏益华, 1982年生, 江西省农业科学院农产品质量安全与标准研究所助理研究员 e-mail: weiyihua08@163.com
建立了一种测定畜禽肉类中硒代胱氨酸、 甲基硒代半胱氨酸、 硒代蛋氨酸、 硒酸根和亚硒酸根含量的离子对反相液相色谱-原子荧光光谱分析方法。 样品中有机硒通过胰蛋白酶和蛋白酶(XIV, 链霉蛋白酶)酶解提取, 无机硒通过碘乙酰胺溶液提取, 于55 ℃水浴200 r·min-1振荡提取20 h, 提取液高速离心后再经超滤管离心净化, C18反相色谱柱分离, 30 mmol·L-1磷酸氢二铵、 0.5 mmol·L-1四丁基溴化铵和5% ( V/V)甲醇为流动相, 用20%( V/V)甲酸调节流动相溶液pH至6.0, 离子对反相液相色谱-原子荧光光谱法测定样品溶液中5种硒形态含量。 采用与标准样品对照法定性, 峰面积外标法定量。 保留时间定性, 外标法峰面积定量。 硒代胱氨酸、 甲基硒代半胱氨酸、 硒代蛋氨酸、 硒酸根和亚硒酸根在5~200 μg·L-1范围内线性良好, 相关系数均大于0.999, 其检出限分别为0.89, 0.78, 0.55, 0.94和0.70μg·L-1, 加标回收率为76.8%~109%, 批内精密度和批间精密度分别为2.7%~7.8%和3.5%~12.3%。 本方法具有快速简便、 灵敏和准确等优点, 适用于畜禽肉类样品中硒形态分析测定。
, HUANG Qing-qing, QIU Su-yanA method of ion-pair reversed phased liquid chromatography-ultraviolet (IP-RP-HPLC)-hydride generation-atomic fluorescence spectrometry (AFS) was established to determine the contents of selenocystine, methylselenocysteine, selenotmethionine, selenateand selenite in livestock and poultry meat. Organic Se species contents in samples were extracted by trypsin and protease (XIV, pronase), and inorganic Se species contents in samples were extracted with iodine acetamide solution and water incubated at 55 ℃ with shaking at 200 r·min-1for 20 h. The extraction solution of samples was centrifuged at high speedand then centrifuged with microsep for purification. C18 separated the solution of samples reversed phase column by using 30 mmol·L-1diammonium hydrogen phosphate, 0.5 mmol·L-1tetrabutyl-ammonium bromide and 5% ( V/V) methanol as moblie phase. The pH of the mobile phase was adjusted to 6.0 by 20% ( V/V) formic acid. IP-RP-HPLC-AFS determined the contents of five Se species in samples solution. The impurities are qualitatively determined by contrasting with the standard sample and quantitatively determined by calculating the peak areas. Selenocystine, methylselenocysteine, selenotmethionine, selenate and selenite had good linearities in the range of 5~200 μg·L-1, and the correlation coefficients were all greater than 0.999, the detection limits of five Se species were 0.89, 0.78, 0.55, 0.94 and 0.70 μg·L-1, respectively. There coveries were between 76.8%~109%, the within-run precisions and between-run precisions were 2.7%~6.8% and 3.5%~12.3% respectively. The proposed method has the advantages of rapid, simple, high sensitivity and high accuracy, and it is suitable for Se species analysis in livestock and poultry meat samples.
硒(Se)是人体必需的一种微量元素, 为谷胱甘肽过氧化物酶和硒-P蛋白的重要组成部分, 在体内起着平衡氧化还原氛围的作用。 研究表明, 威胁人类健康和生命的40多种疾病, 如癌症、 心血管病、 肝病、 白内障、 胰脏疾病、 糖尿病和生殖系统疾病等都与人体缺硒有关[1]。 然而, 硒适量有益身体健康, 过量则有害健康。 硒摄入量在缺乏和毒性之间范围很窄, 其推荐摄入量和最高安全摄入量上限分别为成人每天50~200和400 μ g[2]。
食物中硒的营养价值不仅取决于总含量, 更取决于其形态种类, 不同化学形态的硒在人体的吸收、 生物效应、 毒性及防癌作用不同[3]。 食物中的硒来源按其存在的形态, 可分为有机硒和无机硒, 有机硒主要形态有硒代氨基酸、 硒代蛋氨酸、 硒蛋白和硒多糖等, 有机硒易被人体吸收利用, 安全性高; 无机硒主要形态有硒酸盐和亚硒酸盐, 无机硒毒性较大, 生物利用率差, 故开展食品中硒形态分析具有重要意义。
目前, 硒形态分析方法有: 气相色谱-质谱法[4], 液相色谱-质谱法[5], 液相色谱-串联质谱法[6], 液相色谱-电感耦合等离子体质谱法[7, 8]和液相色谱-原子荧光光谱法[9, 10]。 与其他方法相比, 液相色谱-原子荧光光谱法具有仪器购置和运行成本低, 光谱干扰少, 线性范围宽和检出限较低等诸多优点, 其元素形态分析应用越来越多。 液相色谱分离硒形态主要方法为离子交换液相色谱法和离子对反相液相色谱法, 其中己烷磺酸钠[8], 氢氧化四甲基铵[11]、 三氟乙酸[12]、 五氟丙酸[13]和七氟丁酸[14]等离子对试剂用于硒形态分析研究报道较多, 而四丁基溴化铵用于硒形态分析研究报道相对较少。
食品中硒形态分析研究对象主要集中于稻谷[15], 大豆[6]、 蔬菜[5]、 水果[16]和饲料[17]等植物性样品。 动物性样品硒形态分析研究极为少见[8, 18], 其测定方法均为液相色谱-电感耦合等离子体质谱法, 且样品需冷冻干燥。 本研究拟采用液相色谱-原子荧光光谱法对畜禽鲜肉样品中硒代胱氨酸、 甲基硒代半胱氨酸、 硒代蛋氨酸、 硒酸根和硒酸根等5 种硒形态进行测定, 以期为畜禽肉类样品中硒形态分析与研究提供参考。
高效液相色谱(SA 20)-原子荧光光谱仪(AFS 9320, 吉天仪器公司), 台式高速冷冻离心机(CR21N, 日立), pH计(S2, 梅特勒), 微波消解仪(Xpress, CEM), 微控数显电热板(EG 37C, 莱伯泰科), Milli-Q5超纯水系统。
硒代胱氨酸、 甲基硒代半胱氨酸和硒代蛋氨酸标准物质(纯度均为98%, 百灵威公司); 亚硒酸根(GBW10032)和硒酸根(GBW10033)标准溶液均购于中国计量科学研究院; 胰蛋白酶(≥ 2 500 units· mg-1, 百灵威), 蛋白酶XIV(≥ 3.5 units· mg-1, 链霉蛋白酶, Sigma-Aldrich); 磷酸氢二铵(ACS), 四丁基溴化铵(离子对色谱级), 甲醇和甲酸(HPLC), 氢氧化钾、 碘化钾、 盐酸和硝酸(均为GR), 硼氢化钾、 碘乙酰胺、 高氯酸和铁氰化钾(均为AR)均购置于阿拉丁公司。 超滤管(Amicon® Ultra-15, 3KDa, 密理博公司)。
1.2.1 硒代胱氨酸、 甲基硒代半胱氨酸和硒代蛋氨酸的提取与净化
取一定量的畜禽肉鲜样充分粉碎混匀。 称取2.0 g(精确至0.001 g)样品于50 mL离心管中, 加入40 mg胰蛋白酶、 40 mg蛋白酶XIV和10 mL 30 mmol· L-1 磷酸氢二铵溶液(pH 8.0), 涡旋混匀, 于恒温水浴振荡器上55 ℃、 200 r· min-1条件下震荡提取 20 h, 10 000 r· min-1离心10 min, 提取液经滤纸过滤再转移至超滤离心管, 于5 000 r· min-1离心10 min, 经0.45 μ m滤膜过滤, 用液相色谱-原子荧光光谱仪测定样品溶液中硒代胱氨酸、 甲基硒代半胱氨酸和硒代蛋氨酸含量。
1.2.2 硒酸根和亚硒酸根的提取与净化
取一定量的畜禽肉鲜样充分粉碎混匀。 称取样品2.0 g (精确至0.001 g)样品, 置于50 mL离心管中, 加入10 mL磷酸氢二铵缓冲液和20 μ L 0.5 mol· L-1碘乙酰胺, 涡旋混匀, 于恒温水浴锅上55 ℃、 200 r· min-1条件下震荡提取 20 h, 10 000 r· min-1离心10 min, 提取液经滤纸过滤再转移至超滤离心管, 于5 000 r· min-1离心10 min, 经0.45 μ m滤膜过滤, 用液相色谱-原子荧光光谱仪测定样品溶液中硒酸根和亚硒酸根含量。
液相色谱-原子荧光光谱仪条件: (1)液相色谱: 采用C18反相色谱柱(4.6× 250 mm, 5 μ m, Inertsil ODS-3, GL Science), 以30 mmol· L-1 磷酸氢二铵、 0.5 mmol· L-1 四丁基溴化铵和5%(V/V)甲醇为流动相, 用20%(V/V)甲酸调节流动相溶液pH为 6.0, 流动相流速为0.8 mL· min-1, 进样量为100 μ L; (2)在线紫外消解系统: 在线紫外灯消解, 还原剂为0.5%(m/v)氢氧化钾+0.2 %(m/v)碘化钾; (3)原子荧光光谱: 0.5%(m/v)氢氧化钾+2.5%(m/v)硼氢化钾, 载流为10%(V/V)盐酸溶液, 硒空心阴极灯主、 辅极电流和电压分别为100 mA, 50 mA和310 V, 载气和屏蔽气流速350和750 mL· min-1。
采用《GB 5009.93— 2017》中氢化物原子荧光光谱法对样品中总硒进行测定[19]。 取一定量的鸡肉鲜样充分匀浆, 称取2.0 g(精确至0.001 g)样品于锥形瓶中, 加入9 mL硝酸和1 mL高氯酸, 置于电热板上于160 ℃消解4 h, 再升温至260 ℃赶酸至白烟冒尽, 冷却后加入10 mL(V/V)盐酸溶液于95 ℃加热5 min以还原六价硒成四价硒, 消解液转移至25 mL容量瓶中, 加入2.5 mL 10%(m/v)铁氰化钾溶液, 用水定容至刻度。
原子荧光光谱条件: 还原剂为0.5%(m/v)氢氧化钾+2.5%(m/v)硼氢化钾, 载流为10%(V/V)HCl, 硒空心阴极灯主、 辅极电流和电压分别为100 mA, 50 mA和300 V。
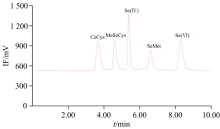
首先, 考察了离子对液相色谱法(IP-RP-HPLC)和阴离子交换液相色谱法(AE-HPLC)对硒代胱氨酸(SeCys2)、 甲基硒代半胱氨酸(MeSeCys)、 硒代蛋氨酸(SeMet)、 硒酸根(Se(Ⅳ ))和硒酸根(Se(Ⅵ ))的分离度和灵敏度的影响。 离子对液相色谱法采用Inertsil ODS-3 C18反相色谱柱, 以30 mmol· L-1磷酸氢二铵、 0.5 mmol· L-1四丁基溴化铵和5% (V/V)甲醇为流动相(pH 6.0), 流速为0.8 mL· min-1, 柱温30 ℃, 见图1; 阴离子交换液相色谱法采用Hamilton PRP-X100阴离子色谱柱, 以40 mmol· L-1磷酸氢二铵和5% (V/V)甲醇为流动相(pH 6.0), 流动相流速为1.0 mL· min-1, 柱温30 ℃。 虽然此两种方法均能成功分离5种硒形态, 但阴离子交换液相色谱法的硒酸根保留时间明显长于离子对液相色谱法的硒酸根保留时间, 且其有机硒(尤其是硒代蛋氨酸灵敏度)较差, 故本研究最终采用离子对液相色谱法分离5种硒形态。
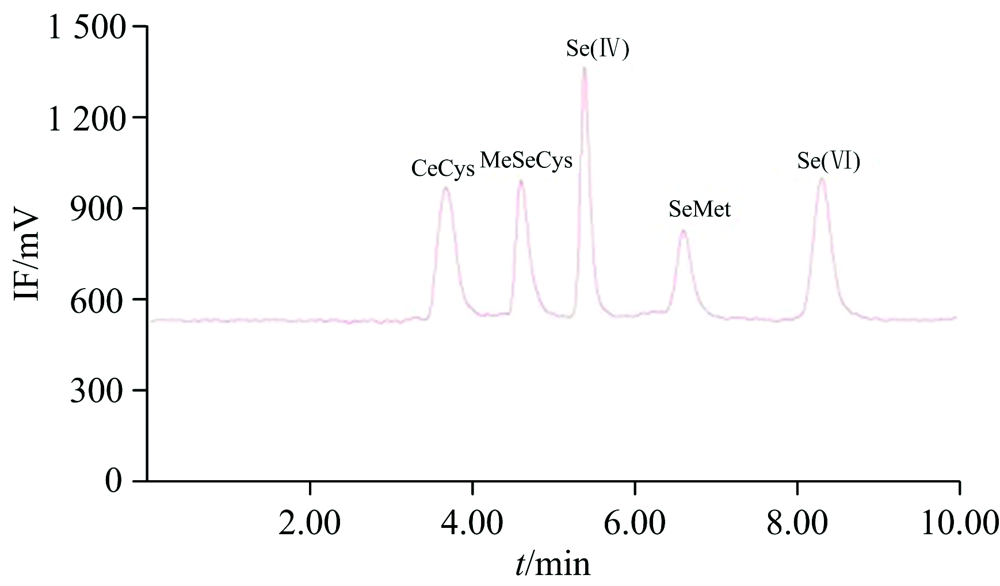 | 图1 IP-RP-HPLC(Inertsil ODS-3色谱柱)硒形态色谱图Fig.1 Chromatogram of Se species with IP-RP-HPLC (Inertsil ODS-3) |
同时, 考察了不同流动相体系对硒形态分离的影响, 共设3种流动相体系, (1) 磷酸盐: 30 mmol· L-1磷酸氢二铵、 0.5 mmol· L-1 四丁基溴化铵和5%(V/V)甲醇(pH 6.0); (2) 乙酸铵: 25 mmol· L-1乙酸铵、 0.1 %(m/v)TFA和5% (V/V)甲醇为流动相(pH 6.0); (3) 柠檬酸: 5 mmol· L-1柠檬酸(pH 4.5)。 磷酸盐和乙酸铵流动相体系均可较好的分离5种硒形态, 分离度和灵敏度亦较佳, 但乙酸铵流动相体系的硒酸根出峰时间相对较晚, 柠檬酸流动相体系的硒代胱氨酸和甲基硒代半胱氨酸分离较差。
此外, 还考察了磷酸盐流动相体系中磷酸氢二铵浓度、 四丁基溴化铵浓度、 甲醇含量和色谱柱柱温对硒形态保留时间和灵敏度的影响。 实验表明, 磷酸氢二铵浓度、 四丁基溴化铵浓度、 甲醇含量和pH对硒酸根保留时间影响较大, 而对有机硒保留时间影响较小。 磷酸氢二铵浓度和甲醇含量的增加会明显缩短硒酸根保留时间, 而四丁基溴化铵浓度增加会明显延长硒酸根保留时间。 甲醇含量的增加可显著提高硒形态灵敏度。 色谱柱柱温对硒形态保留时间和灵敏度几乎无影响。
综上所述, 最终流动相体系为30 mmol· L-1磷酸氢二铵、 0.5 mmol· L-1四丁基溴化铵, 5% (V/V)甲醇含量(pH为6.0), 流速0.8 mL· min-1, 5种硒形态Se分离度好, 在10 min之内可完成5种硒形态分析测试工作。
通过在线紫外灯辐射有机硒转化成六价硒, 还原剂碘化钾将六价硒还原成四价硒, 硼氢化钾将四价硒在盐酸介质中还原成硒化氢。 值得注意的是, 即使在紫外消解系统和碘化钾还原剂共同的作用下, 离子对反相液相色谱-紫外-氢化物发生-原子荧光光谱法测定硒形态, 相同浓度的各种硒形态峰面积存在明显差异, 其峰面积从大至小一般为: 亚硒酸根> 硒酸根> 甲基硒代半胱氨酸> 硒代胱氨酸> 硒代蛋氨酸, 其差异原因可能为在线紫外消解系统对各种硒形态转化率不同所致。
本研究对还原剂碘化钾、 硼氢化钾以及载流(盐酸)浓度等进行优化, 以期达到较佳的灵敏度。 结果表明, 当碘化钾浓度为0.2%, 硼氢化钾浓度为2.5%, 盐酸浓度为10%时, 硒形态灵敏度较高。
2.2.1 蛋白酶种类与用量的选择
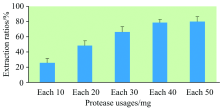
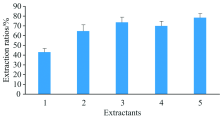
首先, 考察了不同蛋白酶对猪肉样品中硒形态提取效果, 共设5个蛋白酶处理, 见图2。 除胃蛋白酶处理提取剂为0.1(V/V)%盐酸(pH 3.0), 其他处理提取剂均为30 mmol· L-1 磷酸氢二铵溶液(pH 8.0), 提取方式为水浴震荡200 r· min-1 20 h。 样品提取率等于样品各种硒形态含量之和除以样品总硒含量之和。 实验表明, 蛋白酶复合处理提取效果明显高于单一蛋白酶处理, 胰蛋白酶+蛋白酶XIV提取效果最佳, 其次为蛋白酶XIV+脂肪酶, 胃蛋白酶提取效果最差。 故本实验最终选择胰蛋白酶+蛋白酶XIV对样品中硒形态进行提取。 其次, 对胰蛋白酶+蛋白酶XIV用量进行了考察, 共设5个处理, 见图3。 实验表明, 40 mg胰蛋白酶+40 mg蛋白酶XIV、 50 mg胰蛋白酶+50 mg蛋白酶XIV对样品硒形态提取率较高, 二者提取率无显著差异。 但由于蛋白酶价格较高, 选取40 mg胰蛋白酶+40 mg蛋白酶XIV用量有利于降低试剂成本。
 | 图2 不同蛋白酶对样品硒形态提取率的影响(n=3)Fig.2 Influences of extraction rates of Se species in samples with different proteases (n=3) |
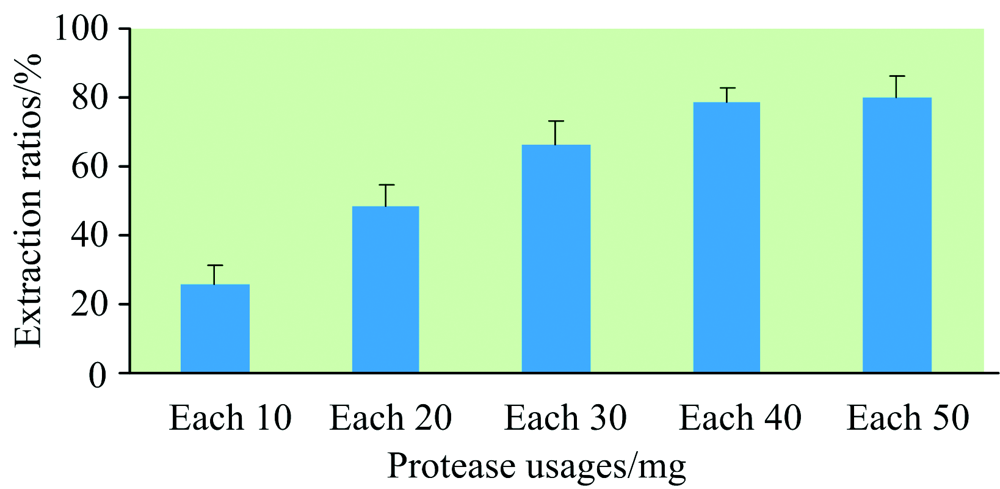 | 图3 蛋白酶(胰蛋白酶、 蛋白酶XIV)不同用量对样品硒形态提取率的影响Fig.3 Influences of extraction rates of Se species in samples with different dosage of proteases (trypsin, protease XIV) |
2.2.2 样品提取方式及提取时间的选择
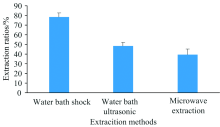
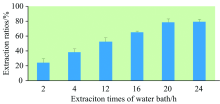
首先, 考察了水浴震荡提取、 水浴超声提取和微波提取对样品硒形态提取效果的影响。 水浴震荡提取条件为55 ℃水浴振荡器上200 r· min-1提取24 h, 超声提取条件为37 ℃超声30 min× 4次(频率为20 kHz), 微波提取条件为40 ℃微波30 min(微波功率为100 W)。 从图4可知, 样品硒形态提取率从大至小为: 水浴震荡提取> 水浴超声提取> 微波提取。 同时进行添加回收试验, 添加硒形态种类为硒代胱氨酸、 甲基硒代半胱氨酸和硒代蛋氨酸以考察提取方式是否会破坏有机硒硒形态结构。 结果表明, 水浴提取下有机硒形态未发生转化, 超声提取时硒代胱氨酸峰附近出现未知物质峰, 估计为长时间超声导致硒代胱氨酸形态发生转化所致, 微波提取下有机硒硒形态未发生转化, 但其提取率较低。 故本研究最终选用水浴震荡提取方式对样品中硒形态进行提取。 其次, 考察了水浴震荡提取时间对样品硒形态提取率的影响, 共设6个处理。 从图5可知, 当水浴震荡提取时间≥ 20 h, 样品硒形态提取率较好, 故最终水浴震荡提取时间设为20 h。
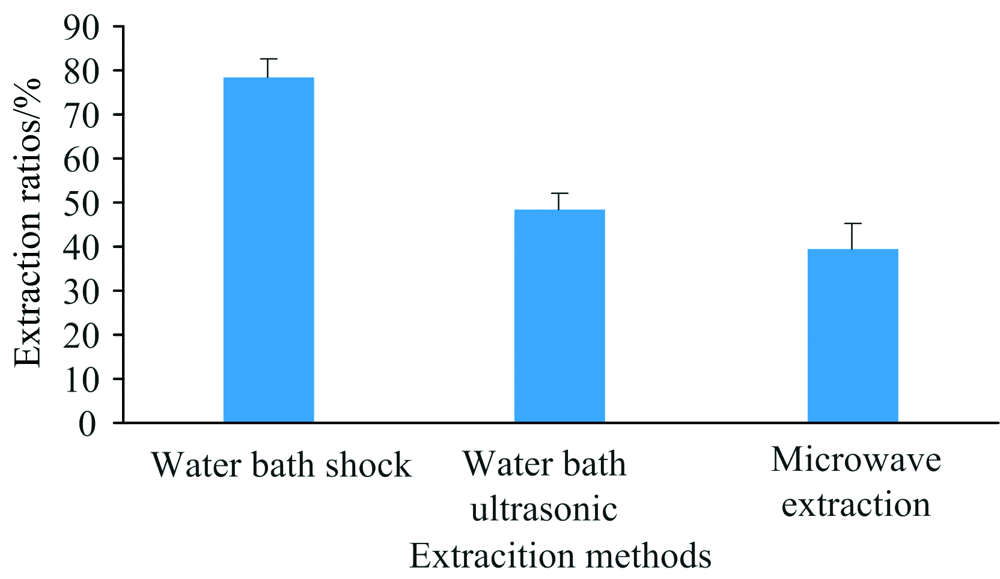 | 图4 不同提取方式对样品硒形态提取率的影响(n=3)Fig.4 Influences of extraction rates of Se species in samples with different extraction means (n=3) |
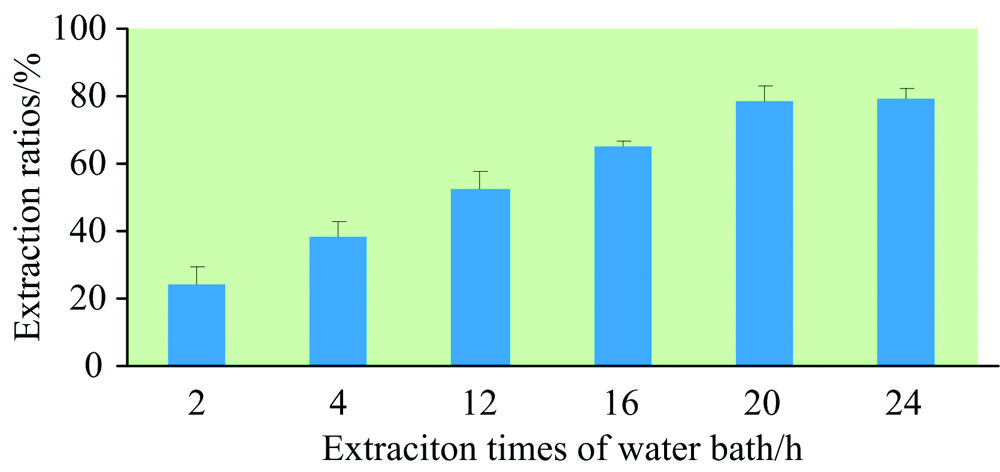 | 图5 不同水浴提取时间对样品硒形态提取率的影响(n=3)Fig.5 Influences of extraction rates of Se species in samples with different extraction times by water bath (n=3) |
2.2.3 样品提取剂的选择
由于胰蛋白酶和蛋白酶XIV均为碱性蛋白酶, 提取体系中pH值对酶活力影响较大。 三(羟甲基)氨基甲烷(Tris-HCl)和磷酸氢二铵[(NH4)2HPO4]均为常见缓冲液, 用作提取剂可起稳定pH作用, 本实验考察了不同提取剂及pH值对样品硒形态提取率的影响, 共设5个处理, (1)水、 (2) 30 mmol· L-1 Tris-HCl(pH 7.5)、 (3) 30 mmol· L-1 Tris-HCl(pH8.0)、 (4) 30 mmol· L-1(NH4)2HPO4(pH7.5)和(5) 30 mmol· L-1 (NH4)2HPO4(pH 8.0)。 从图6可知, 样品硒形态提取率从大至小顺序为30 mmol· L-1 (NH4)2HPO4(pH 8.0)> 30 mmol· L-1 Tris-HCl(pH8.0)> 30 mmol· L-1 (NH4)2HPO4(pH 7.5)> 30 mmol· L-1 Tris-HCl(pH 7.5)> 水, 故本方法采用30 mmol· L-1 (NH4)2HPO4(pH 8.0)为样品提取剂。
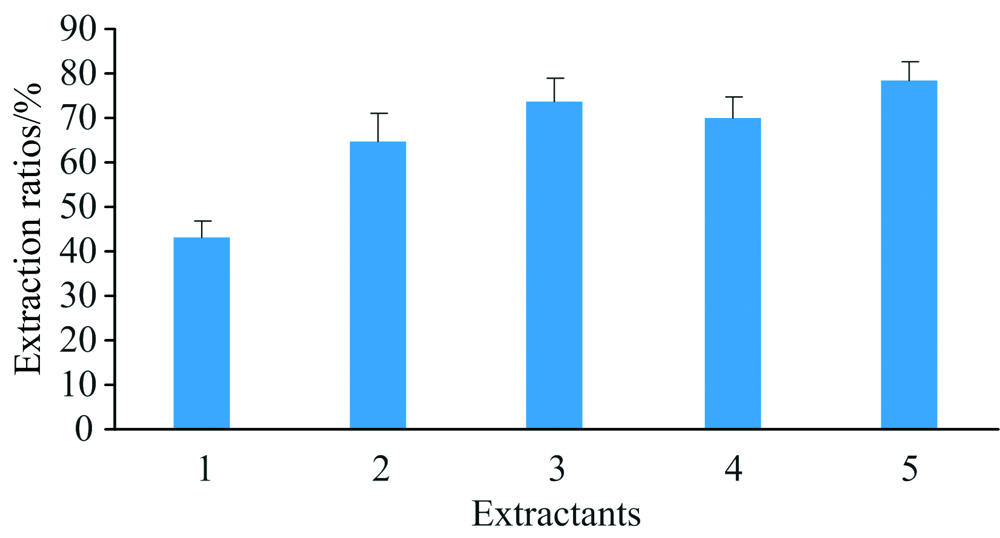 | 图6 不同提取剂对样品硒形态提取率的影响Fig.6 Influences of extraction rates of Se species in samples with different extraction reagents |
2.2.4 样品净化方式的选择
样品经蛋白酶水解后, 溶液颜色较深, 含有较多水溶性蛋白质等杂质会严重缩短色谱柱的使用使命, 故本研究考察了不同净化方式对样品溶液颜色的影响以及对硒形态添加回收率的影响。 共设4个处理, 样品提取液未净化; 样品提取液经超滤管(3KD)离心净化; C18基质固相分散净化, 即提取前往样品中加入0.2 g C18粉末, 其他步骤相同; PSA基质固相分散净化, 即提取前往样品中加入0.2 g PSA粉末, 其他步骤相同。 实验表明, 样品净化后溶液颜色从深至浅依次为: 未净化> C18> PSA≈ 超滤, 4种净化方式的有机硒硒形态添加回收率无明显差异, 但PSA会降低无机硒回收率, 故本方法最终采用超滤对样品溶液进行净化。
2.2.5 碘乙酰胺的作用
在添加回收实验过程中, 本研究发现: 样品经蛋白酶水解, 其亚硒酸根回收率非常低, 通常为0%~5%, 而其他4种硒形态回收率在80%~110%之间。 即使样品不加蛋白酶水解, 只用水或磷酸氢二铵溶液提取, 亚硒酸根回收率亦呈同样结果, 其原因可能为亚硒酸根吸附在样品颗粒表面, 未能解吸到样品溶液中。 通过本研究发现, 样品提取前加入碘乙酰胺溶液, 亚硒酸根回收率显著升至80%以上, 其原因可能为碘乙酰胺破坏了蛋白结构或其还原性使亚硒酸根解吸到样品溶液中。
由于碘乙酰胺是一种氨基酸烷基化试剂, 能与硒代胱氨酸、 硒代蛋氨酸等有机硒发生烷基化反应。 添加回收实验亦证明, 当碘乙酰胺存在时, 硒代蛋氨酸和硒代胱氨酸结构发生转化, 其回收率明显下降, 故同时测定畜禽肉中5种硒形态, 样品提取步骤需要分成两步, 第一步为样品中有机硒的提取, 其提取需经蛋白酶水解; 第二步为样品中无机硒的提取, 其提取仅需加入磷酸氢二铵溶液, 无须酶解。
2.3.1 样品基体效应的考察
通过往样品提取液中添加一定量硒形态标准溶液以考察样品基质效应(ME)。 样品基质效应=100%× (样品添加浓度的荧光强度-样品本底的荧光强度)/标准溶液浓度的荧光强度, 假设80%≤ 样品基质效应≤ 120%, 则认为样品的基质效应较小, 用标准曲线法分析测试即可; 若样品基质效应< 80%或> 120%, 则认为样品的基质效应较大, 必须采用标准加入法。 从表1可知, 样品中5种硒形态基质效应为83.7%~119%, 表明其基体效应较小, 用普通标准曲线法定量即可。 由于本研究采用的是离子对液相色谱法, 样品提取剂为30 mmol· L-1 磷酸氢二铵(pH 8.0), 故标准溶液亦须用30 mmol· L-1 磷酸氢二铵(pH 8.0)进行稀释与定容。
| 表1 样品的基质效应 Table 1 Matrix effects of the 5 kinds of Se species in samples |
2.3.2 添加回收率和精密度
通过添加回收率试验以考察方法的添加回收率、 批内精密度和批间精密度, 精密度用相对标准偏差(RSD)。 添加回收试验, 共设置低、 中、 高3个浓度水平, 每平行设6个重复, 批内精密度为同一批次的测量结果, 批间精密度为不同批次(3次)的测量结果。 从表2可知, 本方法5种硒形态添加回收率为76.8%~109%, 批内精密度和批间精密度分别为2.7%~7.8%和3.5%~12.3%。
| 表2 猪肉中5种硒形态的加标回收率和精密度(n=6) Table 2 Standard recovery rates and precisions of 5 kinds of Se species in pork samples (n=6) |
2.3.3 方法线性和检出限
从表3可见, 各种硒形态在5~200 μ g· L-1范围内线性良好, 其相关系数均大于0.999。 以信噪比S/N=3为检出限, 5种硒形态检出限为0.55~0.94 μ g· L-1。 与其他文献相比, 本方法灵敏度与HPLC-ICP-MS, HPLC-MS/MS灵敏度相近, 高于HPLC-TR-HG-AFS灵敏度, 且该方法测定硒形态种类较多[7, 16, 19]。
| 表3 5种硒形态的线性范围、 线性方程、 相关系数和检出限 Table 3 Linear range, linear equation, correlation coefficients and detection limits |
测定畜禽肉样品中总硒含量和硒形态含量, 以观察本方法对样品中硒形态的提取率。 提取率等于样品各硒形态含量之和除以样品总硒含量。 从表4可知, 本方法样品硒形态提取率较高, 富硒畜禽肉中主要形态为硒代蛋氨酸和硒代胱氨酸, 未发现无机硒。
| 表4 样品硒形态和总硒含量的测定(n=3) Table 4 Determination of Se species and total Se contents in samples(n=3) |
建立了一种用离子对液相色谱-原子荧光光谱法测定硒代胱氨酸、 甲基硒代半胱氨酸、 硒代蛋氨酸、 硒酸根和亚硒酸根5种硒形态含量的方法。 畜禽肉类样品无须冷冻干燥处理, 鲜样样品中有机硒经胰蛋白酶和蛋白酶XIV酶解水浴提取, 无机硒经碘乙酰胺溶液提取, 以磷酸盐体系为流动相, 四丁基溴化铵为离子对试剂, 经C18反相色谱柱分离, 10 min内可以完全分离5种硒形态。 本方法较为简便、 灵敏和准确, 为畜禽肉类硒形态分析与研究提供了切实可行的分析方法和技术支撑。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|