


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
联合特征子空间分布对齐的标定迁移方法
[赵煜辉 , 刘晓东, 张磊, 刘永宏]
, 刘晓东, 张磊, 刘永宏]
, 刘晓东, 张磊, 刘永宏]
|
|


作者简介: 赵煜辉, 1971年生, 东北大学秦皇岛分校教授 e-mail: 1000272@neuq.edu.cn
近红外光谱分析技术近年来在各种领域的定性、 定量分析等方面得到广泛的应用。 多元标定技术则是光谱分析领域中最先进的技术, 而环境条件、 测量仪器或测量物质自身的变化, 都可能导致多元标定模型不再适用于新样本的预测。 重新标定和重新建模必然会浪费大量时间和资源。 一种解决方案是标定迁移, 将源域已有的标定模型扩展到目标域中, 避免重复建模的代价。 在化学计量学的相关文献中, 绝大多数迁移方法都需要在两台仪器相同条件下都测量一组迁移标准样品, 但在近红外光谱测量技术中, 由于标准样品具有挥发等特性, 使得构建仪器标定迁移方法的标准样品难以获得和保存。 针对这些问题, 提出了一种联合特征子空间分布对齐(JSDA)的标定迁移方法, 此方法可以在从仪器没有标准样本的情况下建立标定迁移模型。 JSDA首先建立源域和目标域数据特征的联合主成分分析(PCA)子空间; 然后通过对齐映射在联合特征子空间中的源域特征分布和目标域特征分布来校正标定模型; 最后, 应用最小二乘模型构建校正后源域上的标定模型, 该模型可直接用于目标域的标定。 实验结果表明与已有成熟的标定迁移方法相比, JSDA在公开的真实数据集上的预测性能比较有优势, 验证了该模型在实际应用中的有效性和优越性。
Near-infrared spectroscopy analysis technology has the advantages of low cost, high efficiency, and pollution-free. In recent years, it has been widely used in qualitative and quantitative analysis in various fields. Multivariate calibration technology is the most advanced technology in the field of spectroscopy. Changes in conditions, instruments, or substances may cause the multivariate calibration model to no longer be suitable for the prediction purposes of newly measured samples. Re-calibration and re-modeling will inevitably waste a lot of time and resources; another option is calibration transfer, which extends the existing calibration model in the source domain to the target domain to avoid the cost of repeated modeling. In the related chemometrics literature, most transfer methods need to measure a set of transfer standard samples under the same conditions of two instruments. However, in the near-infrared spectroscopy measurement technology, due to the characteristics of volatilization of the standard samples, It is not easy to obtain and save the standard samples for constructing the transfer method for instrument calibration. This paper proposes a joint feature subspace distribution alignment (JSDA) calibration transfer method in response to these problems. This method can establish a calibration transfer model without a standard sample from the instrument. JSDA first establishes the joint PCA subspace (Principal component analysis) of the data features of the source and target domains; then corrects the calibration model by aligning the source domain feature distribution and target domain feature distribution mapped in the joint feature subspace; Finally, the least squares model is used to build a calibration model on the corrected source domain, which can be directly used for the calibration of the target domain. The experimental results show that compared with the existing mature calibration transfer methods, JSDA has more advantages in predicting performance on public real data sets, which verifies the effectiveness and superiority of the model in practical applications.
近红外光(NIR)是一种波长在780~2 526 nm之间的电磁波, 近红外光谱区与有机分子中含氢基团(O—H, N—H, C—H)振动的合频和各级倍频的吸收区一致, 通过扫描样品的近红外光谱, 可以得到样品中有机分子含氢基团的特征信息[1, 2]。 近红外光谱的多元标定方法是利用含有氢基团化学键伸缩振动倍频和合频, 在近红外区域的吸收光谱, 通过选择适当的化学计量学领域的多元标定方法, 找到标定样本的近红外吸收光谱与其相应的成分浓度或性质数据之间的关联, 建立两者之间的标定关系模型[3]。 主成分回归(principal component regression, PCR)[4]和偏最小二乘(partial least squares, PLS)[5]等标定方法已经被证实是有效的, 建立可靠的多元标定模型通常耗时且成本高昂, 然而在实际工业生产中, 通过对原有近红外光谱数据进行分析建立的模型往往对新的数据集并不适用, 从而导致原有模型失效。 解决此类问题通常有两种方法: 一是重新对新的数据集进行重新标定和重建模型; 二是建立标定迁移模型, 将已有可靠的源域多元标定模型迁移到目标域中。 重新标定和重建模型需要耗费大量的时间和资源[6], 而标定迁移不仅可以有效的避免这一缺点, 而且还可以使得目标领域取得可靠的学习效果。 显然, 选择第二种方法是解决此类问题的最佳策略[7]。
一般来说, 标定迁移方法可以分为两类: 有标样的标定迁移和无标样的标定迁移。 目前比较有代表性的有标样的标定迁移方法有直接标准化(direct standardization, DS)[8]、 分段直接标准化(piecewise direct standardization, PDS)[9]、 基于典型相关分析的标定迁移(canonical correlation analysis based calibration transfer, CCACT)[10, 11]以及斜率和偏差校正算法(slope bias correction, SBC)[12]等, 无标样的标定迁移方法有多元散射校正(multiplicative scatter correction, MSC)[13]、 迁移成分回归(transfer component regression, TCR)[14]等, 其中DS和PDS的前提是假设光谱响应的变异都是测量环境引起的; 但是实际上, 我们所收集和整理的化学样品也存在着一定的不确定性; SBC为一种单变量方法, 因此在测量仪器和测量条件变化引起系统化的光谱差异的情况下, 才能取得较好的效果。 现实生活中, 光谱差异往往比较复杂, 此时它的预测能力是不确定的; MSC 预处理方法并不能显著提高模型的预测能力; TCR虽然具有较好的泛化能力, 但与其他方法相比预测精度较低。
大多数能够显著地提高预测性能的迁移方法都属于有标样的标定迁移方法, 即需要标准样本来构建标定迁移模型, 且标准样本中主仪器与从仪器的样本必须一一对应紧密匹配, 具备良好的代表性和适应性, 能够很好地解释两种仪器之间的差异。 由于这些要求的限制, 有标样的模型通常泛化能力较差。 而已被提出的少量无标准标定迁移方法虽然不需要标准样本, 但其预测性能与有标样的标定迁移方法相比相差较大。 因此, 结合两者优点, 开发一种性能可与有标样的迁移方法媲美的无标准样本的迁移学习方法, 将具有很大的意义。 因此结合近红外光谱维度高且存在多重共线性的特点, 以主成分回归(PCR)作为标定模型, 应用迁移学习的思想, 提出了一种无标准样本的基于联合特征子空间分布对齐(joint feature subspace distribution alignment, JSDA)的标定迁移方法, 在不需要标准样本的情况下, 取得相同甚至优于已有经典有标样的标定迁移方法的预测性能。
在本文中, 源域和目标域将各用下标“ s” 和“ t” 表示, Xs=[
下面我们将具体说明如何建立基于近红外光谱特征预测物质成分浓度的无标准样本的标定迁移模型。 用均值和协方差来描述光谱数据分布。 由于均值在数据预处理(如中心化)后通常为零, 不受子空间投影的影响, 因此不需要对它们进行处理。 协方差反映着多维空间基向量之间的相关关系, 源域和目标域的协方差矩阵存在差异, 且向子空间投影会对其产生影响, 因此我们需要消除投影后两者特征光谱协方差矩阵之间的差异, 进而使得两者数据分布对齐[15]。
下面我们从理论上详细阐述JSDA模型的建立过程:
第一步: 构建联合公共特征子空间
对于光谱数据, 一般均为高维小样本X∈ Rn×m, m> n, 即光谱属性维度远大于样本个数。 针对此类数据, 为了便于构建预测模型, 一般要对其进行降维, 如PCA, 将高维样本空间X投影转换为低维子空间Φ ∈ Rn×d, n> d。 标定迁移问题中, 我们可以利用一种特殊的方式直接构建一个公共特征子空间, 将源域和目标域数据均投影到该子空间中, 如此, 两者之间不存在子空间漂移的问题。 对于属性维度相同的光谱数据Xs∈
利用PCA构建子空间的过程中涉及到特征值个数d的选取, 可以通过碎石图准则、 累计方差贡献率法等方法进行确定, 即U=[U1, …, Ud, …,
对于传统的子空间对齐方法, 源域与目标域数据分别构建低维特征子空间时, 存在一个问题, 由于投影矩阵Us和Ut的不同, 造成转换后两者特征子空间基存在差异; 通过计算线性映射矩阵来对齐子空间, 从而最小化它们之间分布差异, 这种方法称为子空间对齐。 而我们提出的构建源域和目标域的联合特征子空间, 使得源域和目标域的特征光谱不仅具有相同的子空间基, 并且能够尽可能的保证原始数据在投影到该子空间上的时候不会失真, 达到最优状态, 因此不需要进一步对齐子空间, 又有很好的优越性。
第二步: 特征分布对齐
公共特征子空间中, 源域和目标域具有相同的子空间基, 但这并不能解决两者数据特征分布之间的差异, 不能满足预测模型应用的独立同分布条件。 如上所述, 我们用均值和方差描述一个分布。 前面提到, 均值在数据中心化处理后不受子空间投影的影响, 因此我们只需消除投影后两者特征光谱的协方差差异。 为了最小化源域特征和目标域特征的二阶统计量(协方差: Σ s和Σ t∈ Rd×d)之间的距离, 我们对源域特征进行线性变换A∈ Rd×d, 使用Frobenius范数作为矩阵距离度量, 从而最小化它们之间差异, 如式(2)所示
其中,
进一步对式(2)推导可得
对于半正定的协方差矩阵
而实际应用中根据已有样本估计的光谱数据协方差矩阵常是不可逆的, 因为样本数据集的特征数总大于样本数, 但一般样本可以集中于一个低维子空间中, 构建子空间中的特征光谱, 此时一般可逆。 对于协方差矩阵不可逆的情况, 我们将结果修正如式(5)所示
其中“ +” 表示Moore-Penrose伪逆,
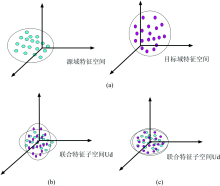
为了便于理解, 我们给出联合特征子空间下的特征分布对齐示意图如图1, 红色表示源域特征样本, 蓝色表示目标域特征样本。 其中图1(a)表示中心化后的两域原始数据投影到联合特征子空间上的分布差异, 图1(b)表示对源域特征进行线性变换后差异。 可以看到经过均值和协方差校正后, 两域的特征分布基本相同。
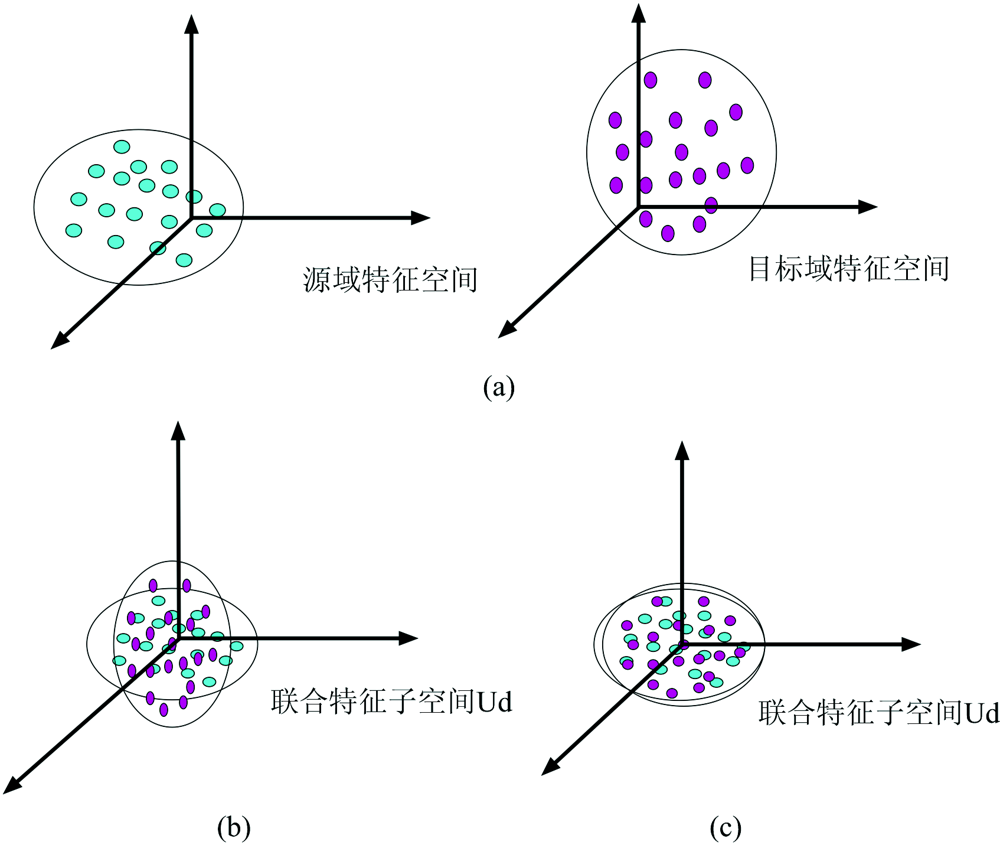 | 图1 特征分布对齐示意图 (a): 原数据; (b)均值校正; (c): 协方差校正Fig.1 Feature distribution alignment diagram (a): Original; (b): Mean correction; (c): Covariance correction |
第三步: 构建目标函数
本工作所解决的标定迁移问题是一个预测问题, 根据上述步骤的结果, 我们可以应用最小二乘法构建校正分布差异后的源域回归预测模型的目标函数, 其形式化如式(6)所示
其中, Φ s为源域投影至公共特征子空间中的特征光谱, A为源域与目标域分布对齐的线性变换矩阵。 β 和b为源域最小二乘回归模型参数的向量形式, 其中β 为回归系数向量, b表示由截距常数b=
经过上述步骤, 源域和目标域具有相同的子空间基, 且实现数据分布对齐, 因而源域上构建的回归模型在两域之前满足数据独立同分布条件。 显然, 上述目标函数求解得到的源域回归模型, 可以直接用于目标域上的回归预测。
第四步: 得到目标域标定模型
上一步中, 源域上得到的最小二乘回归模型参数β 和b可以直接用于目标域上的回归预测, 如式(8)所示
算法: JSDA算法
输入: 主仪器光谱矩阵Xs; 主仪器样本物质浓度矩阵ys; 从仪器光谱矩阵Xt。
输出: 标定迁移模型f(β , b, A)。
开始:
(1)数据中心化处理
(2)构造联合光谱矩阵
(3)利用式(1)找到公共特征子空间Ud;
(4)求得到两域的特征光谱
(5)利用式(2)求解线性映射矩阵
(6)利用式(6)建立源域标定模型, 得到模型参数β 和b, 返回标定迁移模型。
为了验证算法的准确性和实用性, 使用玉米数据集和小麦数据集作为实验对象, 对数据集进行了数据分析, 来检验JSDA方法的性能。
第一个数据集是玉米数据集, 包含三个 NIR 光谱仪(M5, MP5和 MP6)测得的80个样品的光谱数据。 这三台不同的红外光谱仪因其工作原理不同, 所以得到的近红外光谱略有差异, 但对绝大多数谷物而言, 仪器的工作原理不同所产生的误差并不会影响试验结果, 所以我们采用这三台仪器测量的80个玉米的近红外光谱做分析。 玉米数据集中每个样品含有四种成分: 水分, 油, 蛋白质和淀粉。 波长范围为1 100~2 498 nm(700通道), 间隔为2 nm。 该数据集可以从http://www.eigenvector.com/Data/Corn/下载。 仪器M5和仪器MP5之间的光谱差异如图2(a)所示; 仪器M5和仪器MP6之间的光谱差异如图2(b)所示; 仪器MP5和仪器MP6之间的光谱差异如图2(c)所示。 其中横轴表示波长, 纵轴表示吸光度差异(即两种仪器的吸光度差值), 每条曲线代表一个光谱样本。
 | 图2 不用仪器之间的光谱差异Fig.2 Spectral differences between different instruments |
第二个数据集是小麦数据集, 它被用作2016年国际漫反射会议(IDRC)上发布的“ Shootout” 数据集, 选择蛋白质含量作为属性。 小麦数据集的相关信息访问网址http://www.idrc-chambersburg.org/content.aspx?page_id=22& club_id=409746& module_id=191116。 它分析了来自三个不同NIR仪器制造商(A1, A2和A3)的248份小麦数据集的样本。 仪器A1和仪器A2之间的光谱差异如图2(d)所示; 仪器A1和仪器A3之间的光谱差异如图2(e)所示; 仪器A2和仪器A3之间的光谱差异如图2(f)所示。
通过Kennard-Stone(KS)算法将玉米数据集的80个样本分成两组: 80%用做标定集的样本, 20%用做测试集的样本; 将小麦数据集的248个样本分成两组: 80%用作标定集的样本, 20%用作测试集的样本。 对于有迁移标准的迁移方法, 使用Kennard-Stone(KS)算法在标定样本上选择若干个标准样品。
在该实验中, 均方根误差(root mean squard error, RMSE)被用作参数选择和模型评估的指标。 RMSE是预测值与真实值偏差的平方与观测次数n比值的平方根, 可表示数据偏离真实值的程度, 其计算方法如式(9)所示
式(9)中, yi为第i个样本的真实值,
玉米数据集包含各仪器样本各80个, 以M5为主仪器, MP5和MP6分别为从仪器以及MP5为主仪器, MP6为从仪器的实验预测误差RMSEP如表1所示。 小麦数据集包含各仪器样本各248个, 以A1为主仪器、 A2和A3分别为从仪器以及A3为主仪器、 A2为从仪器的实验预测误差RMSEP如表2所示。 其中表中有标样的迁移学习模型(SBC, PDS, CCACT)需要迁移标准样本的个数Nstd不能过少也不能过多, 因此, 在[15, 35]的范围内选取标准样本, 以10为增量, 获取不同数量标准样本对模型预测误差的影响。 观察表中的预测误差结果, 总体来说, 本文提出的JSDA方法在六组对比实验中具有最小的预测误差, 最好的预测精度。 在其他五种有标样和无标样标定迁移方法中, 可以发现三种有标样标定迁移方法(SBC, PDS, CCACT)的预测误差都小于无标样标定迁移方法(MSC, TCR)。 有标样方法虽然需要获取标准样本, 增加了模型的应用代价, 但相应的预测精度也得到了提升, 而无标样方法不需要标准样本, 提高了模型的泛化能力和适用性, 但相应的预测精度也受到了影响。 本文提出的JSDA方法, 很好的解决了无标样标定迁移方法预测精度较低的问题, 在具备与标定迁移方法相同甚至更加优异的预测精度的同时, 还具备良好的适用性, 应用代价较低。
| 表1 SBC, PDS, CCACT, MSC, TCR 和 JSDA 六种迁移方法在玉米数据集下的RMSEP Table 1 RMSEP of corn datasets with SBC, PDS, CCACT, MSC, TCR and JSDA |
| 表2 SBC, PDS, CCACT, MSC, TCR和JSDA六种迁移方法在小麦数据集下的RMSEP Table 2 RMSEP of wheat datasets with SBC, PDS, CCACT, MSC, TCR and JSDA |
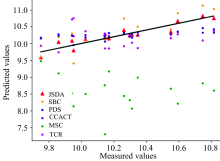
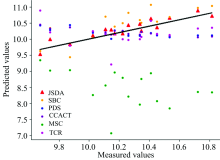
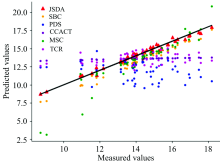
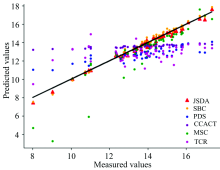
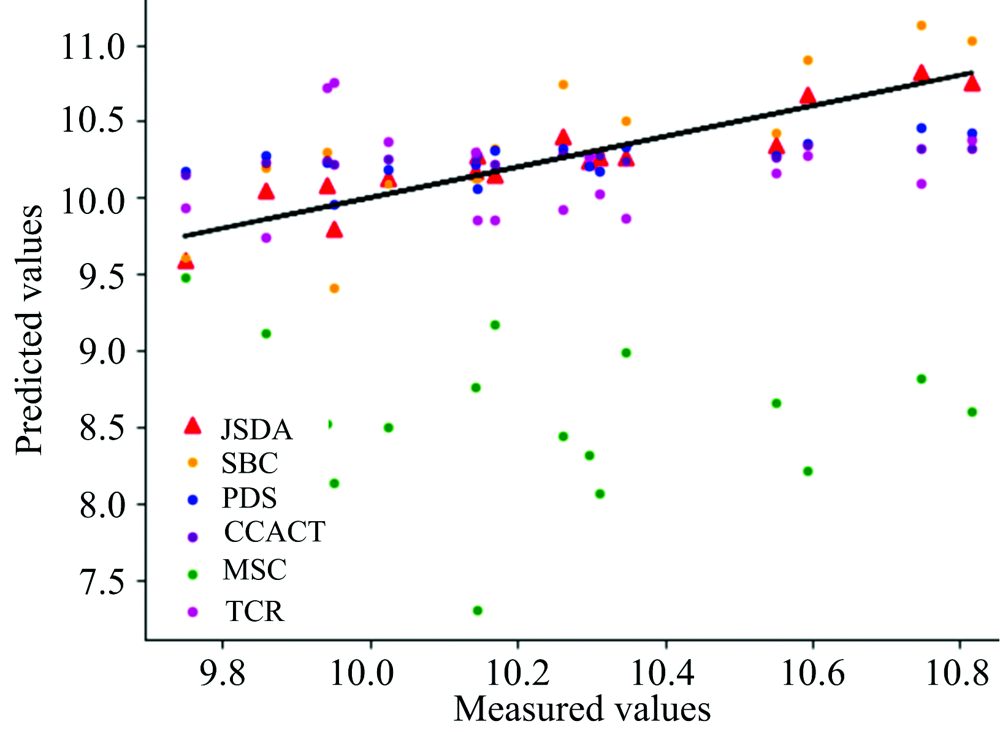
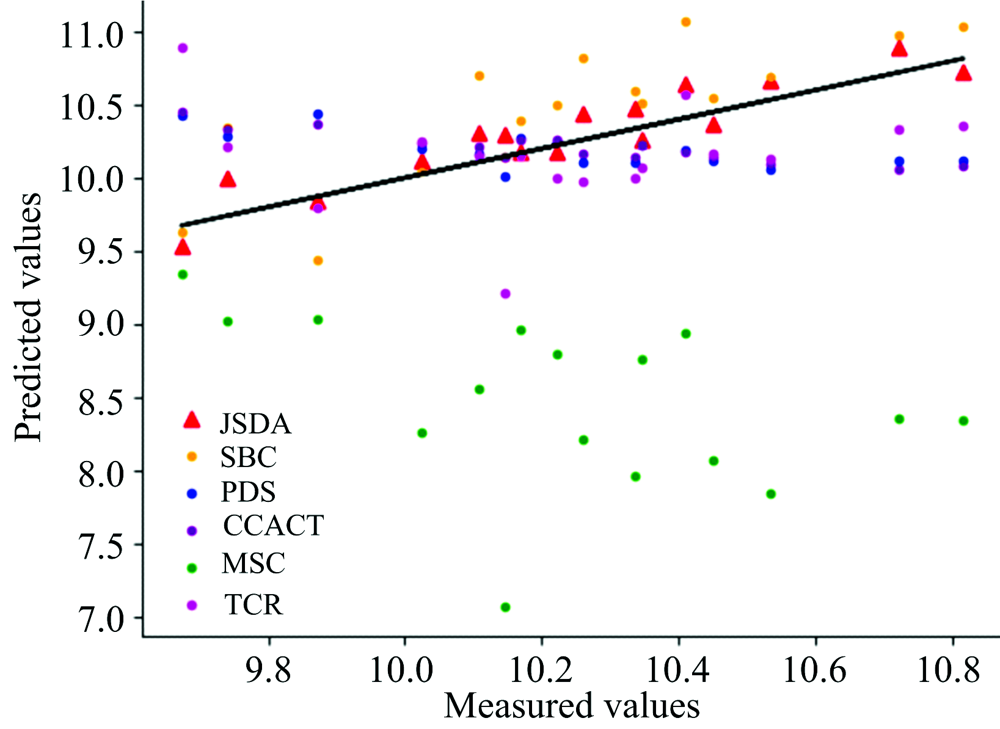
为了直观地观测六种标定迁移方法的性能, 实验中, 以从仪器测试集的物质浓度数据测量值为横坐标, 以标定迁移方法的预测值为纵坐标, 描绘玉米数据集三组实验和小麦数据集三组实验的观测浓度与预测浓度关系图, 如图3—图8所示。 图中的无差异直线表示, 若观测浓度与预测浓度之间误差为零, 则对应的样本点会落在此直线上。 对比观察图3—图8中的预测结果可知, 六种模型中MSC模型在两组实验四种物质上的预测结果基本都聚集在无差异直线的某一侧, 这与表1和表2中展示的结果相呼应, 表明MSC模型的性能较差, 无法准确的标定从仪器的物质浓度。 而CCACT, PDS, SBC, TCR以及本文提出的JSDA模型在两组实验上的预测结果基本都聚集在无差异直线的两侧, 分布都较为均匀, 但相对来说, SBC模型的预测结果分布较为散乱, 表明模型鲁棒性较差。 对比所有模型的预测结果, 以JSDA模型的预测结果最为贴近无差异直线, 拟合效果最好, 结合表1和表2中的结果, 可以得知, 本文提出的JSDA方法具备最佳的预测性能, 同时具有更好的泛化能力。
 | 图3 JSDA, SBC, PDS, CCACT, MSC, TCR六种方法在仪器M5和仪器MP5之间预测结果的散点图Fig.3 Scatter plots of prediction comparison between instruments M5 and MP5 using JSDA, SBC, PDS, CCACT, MSC, TCR |
 | 图4 JSDA, SBC, PDS, CCACT, MSC, TCR六种方法在仪器M5和仪器MP6之间预测结果的散点图Fig.4 Scatter plots of prediction comparsion between instruments M5 and MP6 using JSDA, SBC, PDS, CCACT, MSC, TCR |
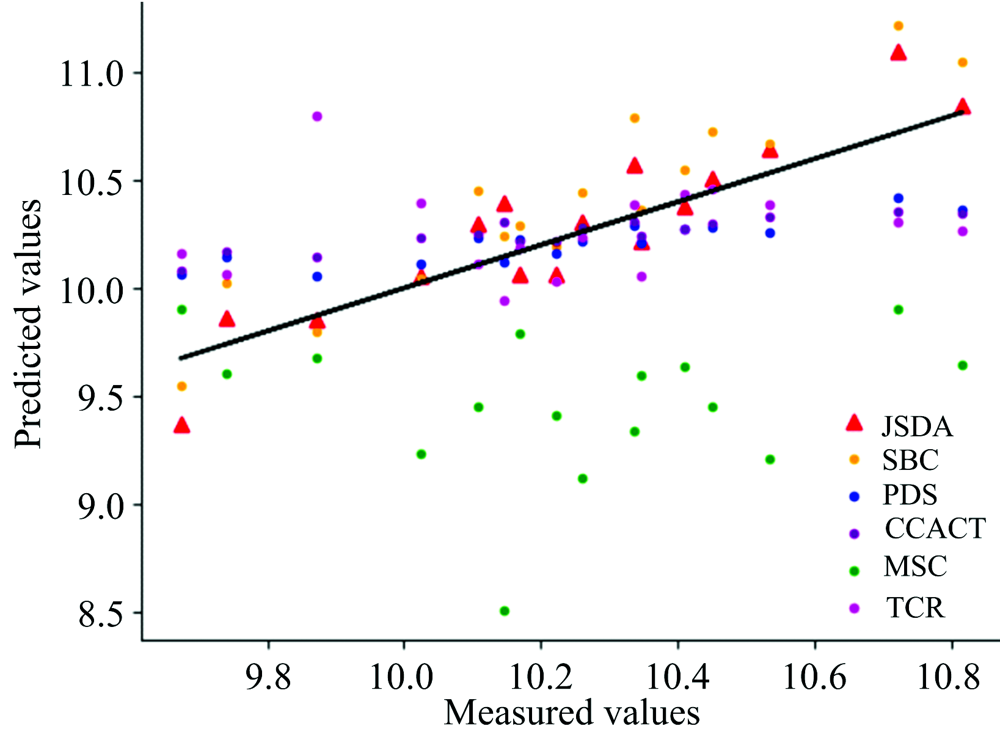 | 图5 JSDA, SBC, PDS, CCACT, MSC, TCR六种方法在仪器MP5和仪器MP6之间预测结果的散点图Fig.5 Scatter plots of prediction comparison between instruments MP5 and MP6 using JSDA, SBC, PDS, CCACT, MSC, TCR |
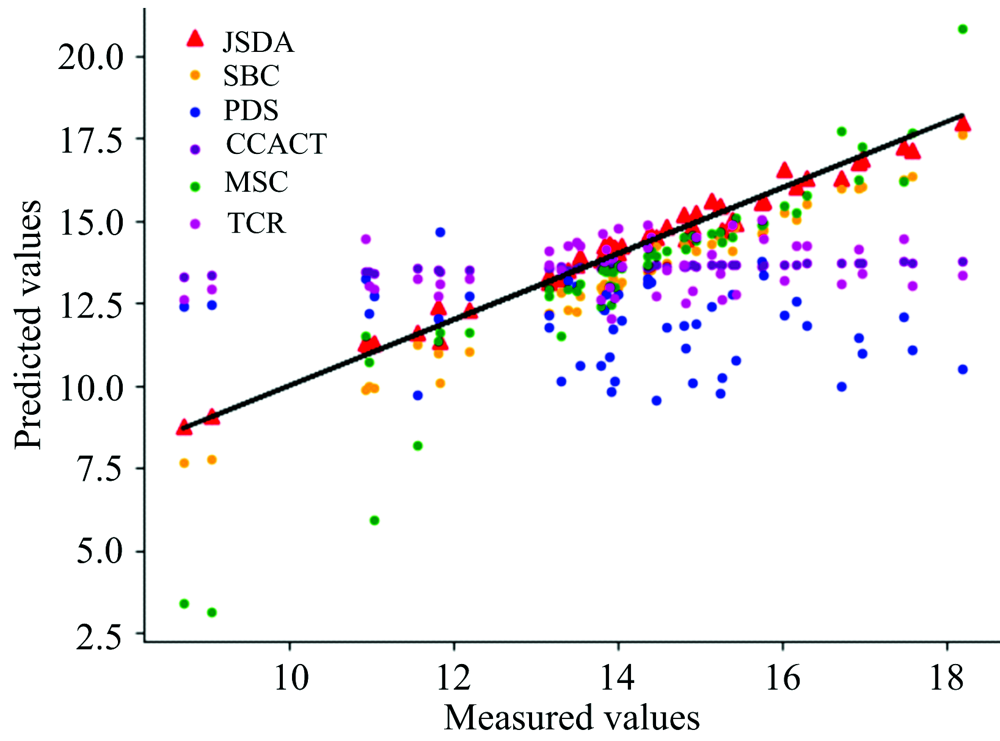 | 图6 JSDA, SBC, PDS, CCACT, MSC, TCR六种方法在仪器A1和仪器A2之间预测结果的散点图Fig.6 Scatter plots of prediction comparison between instruments A1 and A2 using JSDA, SBC, PDS, CCACT, MSC, TCR |
 | 图7 JSDA, SBC, PDS, CCACT, MSC, TCR六种方法在仪器A1和仪器A3之间预测结果的散点图Fig.7 Scatter plots of prediction comparison between instruments A1 and A3 using JSDA, SBC, PDS, CCACT, MSC, TCR |
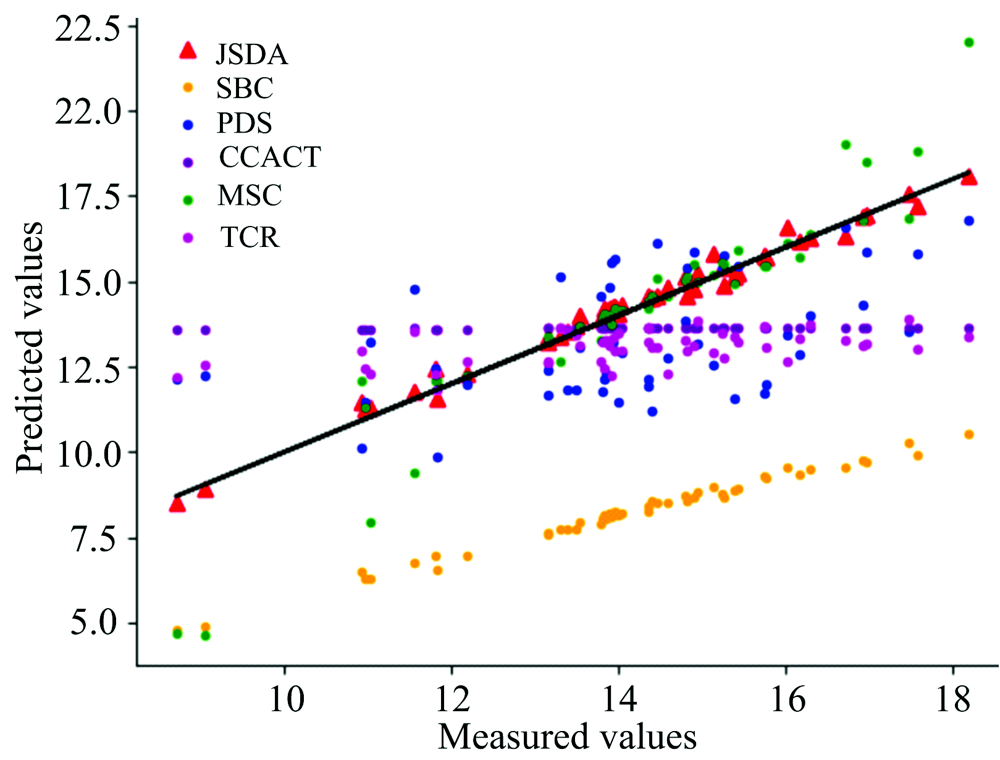 | 图8 JSDA, SBC, PDS, CCACT, MSC, TCR六种方法在仪器A3和仪器A2之间预测结果的散点图Fig.8 Scatter plots of prediction comparison between instruments A3 and A2 using JSDA, SBC, PDS, CCACT, MSC, TCR |
通过在玉米和小麦的近红外光谱数据集上, 在JSDA与SBC, PDS, CCACT, MSC, TCR五种对比标定迁移方法之间, 进行的两组对比实验, 验证了本文方法的性能。 总体来说, 实验结果中, 本文提出的JSDA方法的预测误差都是最低的, 表明在实验的两个数据集上, JSDA方法的性能最优异, 其次是PDS和CCACT, SBC虽然预测的RMSE较小, 但预测结果不稳定, 然后是TCR, 而MSC方法的预测性能最差。 实验结果充分验证了本文所提JSDA方法在实际应用中的优越性, JSDA方法在解决传统标定迁移方法大多需要标准样本这一缺点的同时, 具备与有标样的标定迁移方法相同甚至更优异的性能。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|