
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函理论下多肽光谱性质研究
[杨云帆1  , 胡建波
, 胡建波1 , 刘永刚1, 2, * , 刘强强3 , 张航4 , 徐建洁5 , 郭腾霄5 ]
, 胡建波, 刘强强|
|


作者简介: 杨云帆, 1997年生, 西南科技大学硕士研究生 e-mail: 18281635818@163.com
多肽类物质在生物医药等领域是一种重要的生物大分子, 而紫外-可见吸收光谱和荧光光谱是研究生物分子精细结构的重要手段。 采用密度泛函理论(DFT/RI)计算了生长激素释放肽(GHRP-6)和催产素(Oxytocin)两种多肽的结构模型和分子前线轨道; 在含时密度泛函理论(TDDFT)的基础上, 引入了TDA等近似, 建立了多肽类物质的紫外-可见吸收光谱和荧光光谱的理论模型。 结果表明, 实验测得到GHRP-6的紫外-可见吸收光谱最大吸收波长为279 nm, 计算得到的最大吸收波长为282 nm, 误差为3 nm, 误差百分比约为1%; Oxytocin紫外-可见吸收光谱的实验值为275 nm, 计算值为269 nm, 误差百分比约为2%。 GHRP-6荧光光谱计算值为368 nm, 实验值为360 nm, 误差百分比约为2%; Oxytocin荧光光谱计算值为305 nm, 实验值为312 nm, 误差百分比约为2%。 GHRP-6产生荧光的发射波长与色氨酸产生的荧光波长范围相近, 说明GHRP-6产生荧光的主要贡献为色氨酸残基上的π→π*轨道跃迁, Oxytocin荧光峰位置与酪氨酸产生的荧光波长范围相近, Oxytocin产生荧光的主要贡献为酪氨酸残基上的π→π*轨道跃迁。 根据该模型计算得到的光谱与实验结果吻合度较高, 表明该模型能够准确计算多肽类物质紫外-可见吸收光谱和荧光光谱, 为实验提供可靠的理论依据。
, HU Jian-bo, LIU Qiang-qiangPeptides are important biological molecules. Ultraviolet-visible absorption spectroscopy and fluorescence spectroscopy are important methods for studying the fine structure of biomolecules. The structures and molecular frontier orbital of Growth hormone-releasing peptide (GHRP-6) and Oxytocin were calculated by density functional theory (DFT/RI). Based on time-dependent density functional theory (TDDFT), TDA and other parameter approximations are introduced to establish theoretical models for calculating the UV-Vis and fluorescence spectra of peptides. For GHRP-6, UV spectrum peak is λcal.=282 nm ( λExp.=279 nm, Δ λ=3 nm, Er<2%), and fluorescence spectrum peak is λcal.=368 nm ( λExp.=360 nm, Δ λ=8 nm, Er<3%). For Oxytocin, UV spectrum peak is λcal.=269 nm ( λExp.=275 nm, Δ λ=6 nm, Er<3%), and fluorescence spectrum peak is λcal.=305 nm ( λExp.=312 nm, Δ λ=7 nm, Er<3%). The fluorescence emission wavelength produced by GHRP-6 is similar to the fluorescence wavelength range produced by tryptophan, indicating that the main contribution of GHRP-6 fluorescence is the π*→π orbital transition on the tryptophan residue, Oxytocin fluorescence peak position is similar to the fluorescence wavelength range produced by tyrosine. The main contribution of Oxytocin’s fluorescence is the π*→π orbital transition on tyrosine residues. Calculation results obtain via theoretical models are in good agreement with the experimental. It shows that the models are feasible to accurately calculate the UV Vis absorption spectra and fluorescence spectra of polypeptides, providing reliable theoretical guidance for experiments.
多肽分子是由多种氨基酸残基构成, 其中酪氨酸、 色氨酸、 苯丙氨酸在紫外可见区域有荧光效应[1], 多肽也是蛋白质的组成单元和功能单元, 参与人体的生命活动, 具有出色的生物相容性和化学可修饰性, 因此在纳米药物制备, 荧光探针等领域有着良好的应用前景[2, 3]。 紫外-可见吸收光谱与荧光光谱是研究生物分子的重要手段, 可利用其对分子精细结构进行表征, 在探究不同体系的状态变化, 反应历程等动力学问题时, 起到了重要的作用。
由于泛函的精确度和作用范围的不断提升, 密度泛函理论(DFT)[4]和含时密度泛函理论(TDDFT)[5]成为研究分子基态与激发态性质最常用的方法。 随着电子结构理论研究的不断完善, 模拟计算对激发能(Δ E)和跃迁矩(谱强度)的预测变得越来越准确[6], 计算小体系有机分子的紫外-可见吸收光谱和荧光光谱与实验值的吻合程度越来越高。 国内外对于小体系有机分子的激发态光谱的理论计算做了大量的研究, 通过不同的方法, 在对小体系的激发态进行计算时, 可以拟合出与实验结果吻合度很高的吸收与发射光谱, 国内外的研究者对三种氨基酸的基态构型和荧光光谱进行了理论计算研究; Boo和Ryu等[7]对有机分子Di-9H-fluoren-9-yldimethylsilane的构型对其荧光光谱的影响做了理论计算研究; Dai等[8]对有机分子A100 MOF的荧光光谱和紫外-可见吸收光谱进行了理论计算, 并探究其发光机理。 但当计算的对象体系较大(多肽等生物大分子)时, 通过传统的TDDFT理论来计算其激发态光谱较为困难, 关于激发态性质的密度泛函研究鲜有报道。
采用TDDFT/TDA高精度量子化学计算方法解决TDDFT计算大体系物质激发态困难的问题, 选取多肽模拟物生长激素释放肽-6(GHRP-6)和催产素(Oxytocin)作为较大体系的激发态理论计算研究对象, 通过TDDFT/TDA对多肽的激发态进行计算, 建立了适用于多肽类物质的紫外可见光谱和荧光光谱计算模型, 为较大体系多肽类物质的光谱研究提供参考与借鉴。
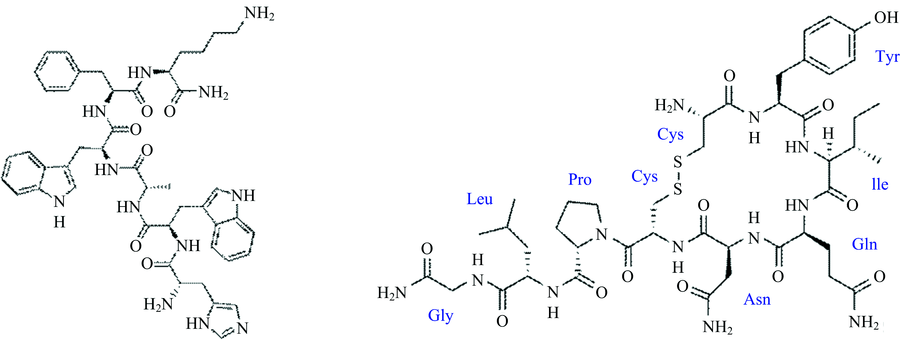 | 图1 生长激素释放肽-6(a)与催产素(b)的2D结构图Fig.1 Structures of GHRP-6 (a), Oxytocin (b) |
采用BP86, B3LYP/def2-SVP(def2/J, def2-SVP/C)计算条件, 为更加充分地考虑分子间弱相互作用, 添加基于BJ阻尼的D3色散矫正[9], 在此条件下优化GHRP-6和Oxytocin的基态几何结构, 并且通过频率分析确定没有虚频, 优化的各收敛限度为: 能量变化(Energy Change): 5.000 0× 10-6 Eh; 最大梯度变化(Max. Gradient): 3.000 0× 10-4 Eh· bohr-1; 均方根梯度变化(RMS Gradient): 1.000 0× 10-4 Eh· bohr-1; 最大位移(Max. Displacement): 4.000 0× 10-3 bohr; 均方根位移(RMS Displacement): 2.000 0× 10-3 bohr。 达到各收敛限度且振动分析无虚频即证明优化完成的结构为分子的最稳定形态。 激发态计算采用相同计算条件, 在不影响计算精度的情况下, 引入RI积分近似和Tamm-Dancoff近似(TDA)来缩短计算时间。 利用该方法对多肽进行几何优化和激发态计算, 以上计算过程在ORCA[10]程序包下进行。
在对激发态性质进行计算时, 当缺乏计算资源或者研究对象的体系较大时, 研究电子跃迁过程将极大的增加计算耗时。 因此对于多肽类物质这种较大体系的分子, 计算其激发态性质时, 采用TDA近似来加速激发态计算过程。 对于完整的TDDFT, 含有非赫米特特征值问题[11],
式(1)中, A, B为旋轨Hessian矩阵, X, Y为特征函数, 为相应的特征值。 由于解出这一问题比较耗时, Hirata and Head-Gordon就提出了Tamm-Dancoff近似, 忽略其中B矩阵的贡献。 将两个特征值问题转化为一个,
由此, X被t取代, ω 被ω TDA取代。 此近似会稍微高估激发能, 但大大减少计算成本, 在对较大体系, 如大多数生物分子进行激发态计算时, 使用此近似可以在节省计算时间的同时保证计算精度, 得到与实验值吻合较高的计算结果。
当光波作用在物质上时, 物质内部的电子将吸收光子能量, 产生从基态跃迁至激发态的电子能级跃迁, 在电子能级跃迁的过程中伴随着振动能级和转动能级跃迁, 因此, 紫外可见吸收光谱为“ 带状光谱” 。 电子能级跃迁所需能量较大, 一般会落在紫外可见区域(200~800 nm)。 电子跃迁符合量子化规则, 因此紫外-可见吸收光谱具有“ 指纹性” 特征, 可以利用紫外-可见吸收光谱对物质做定性定量分析。 研究中对优化完成的多肽的结构进行激发态的单点能计算, 得到与实验值波长范围相同的所有激发态能级的跃迁能, 获得分子的紫外-可见吸收光谱[12]。
光学吸收或发射过程中, 荧光是由自旋允许的单重态到单重态的跃迁发射产生。
分子荧光的强度受分子结构中的刚性平面影响较大, 在组成多肽的多种氨基酸中, 具有荧光效应的主要是三种芳香族氨基酸(色氨酸, 酪氨酸, 苯丙氨酸), 三种氨基酸中均存在的离域π 键, 其荧光主要由π → π * 辐射跃迁产生。 多肽类物质的荧光发射符合Kasha规则, 对于荧光光谱的计算, 通过优化多肽分子第一激发态(S1)的结构, 根据S1结构的分子前线轨道, 预测荧光发射波长的范围; 计算第一激发态到基态的跃迁能, 得到分子的荧光光谱。
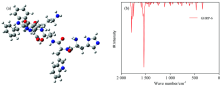
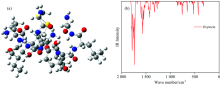
通过DFT方法优化GHRP-6和Oxytocin两种肽气相的几何结构, 优化得到的几何结构见图2(a), 图3(a), 对优化后的结构进行振动分析, 确认没有虚频, 表明该结构为基态最稳定的结构[图2(b)]。 GHRP-6含有6个氨基酸残基, Oxytocin含有9个氨基酸残基。
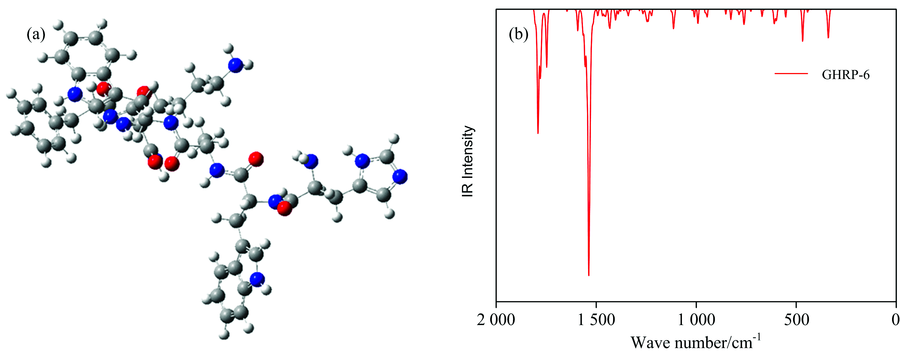 | 图2 GHRP-6优化后的分子结构图(a)和振动光谱图(b)Fig.2 Optimized structures (a) and Vibration spectrum (b) of GHRP-6 |
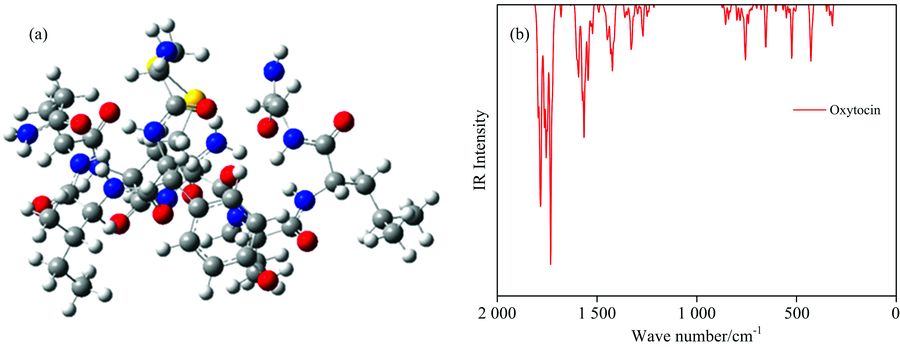 | 图3 催产素优化后的分子结构图与振动光谱图Fig.3 Optimized structures and Vibration spectrum of Oxytocin |
根据优化后的结构, 得到两者多肽中芳香族氨基酸的环与相连原子的二面角, 如表1所示, GHRP-6结构中存在两个荧光贡献基团(色氨酸, TRP), 其二面角更接近平面, Oxytocin上有一个荧光贡献基团(酪氨酸, TYR), 其二面角为14.37° , 与GHRP-6相比, 结构的刚性平面程度和共轭程度更差。
| 表1 多肽结构中芳香族氨基酸与相连原子的二面角 Table 1 Dihedral angles between aromatic amino acids and connected atoms in the polypeptide |
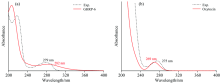
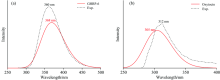
以优化完成后的分子结构为对象, 用TDDFT方法计算两种肽类物质的单重态的激发态跃迁能, 得到两种肽类物质的紫外-可见吸收光谱, 并与实验结果进行比较。 通过对比计算结果, 及其他研究者的工作, 得出了以下结论[6, 13, 14]: 当计算对象的体系越大, 刚性平面程度越低时, 应选取Hartree-Fock(HF)交换成份越高的泛函进行计算。 采用不含HF成份的BP86泛函计算6个氨基酸残基的GHRP-6的紫外-可见吸收光谱与实验值吻合程度最高; 采用HF成份为20%的B3LYP计算9个氨基酸残基的Oxytocin与实验值吻合程度最高。 计算结果见图4(a, b)。
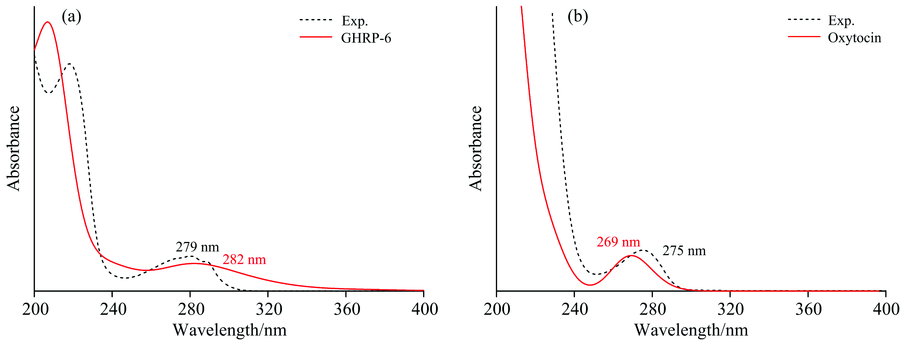 | 图4 GHRP-6(a)和Oxytocin(b)紫外-可见吸收光谱的计算值和实验值Fig.4 UV-Vis spectra of GHRP-6 (a) and Oxytocin (b) Solid: Calculation results; Short Dash: Experimental results |
从图4可以看出, 计算得到的紫外-可见吸收光谱与实验值基本吻合, 实验测得的GHRP-6的紫外-可见吸收光谱最大吸收波长为279 nm, 计算得到的最大吸收波长为282 nm, 误差为3 nm, 误差百分比小于2%[见图4(a)]; Oxytocin紫外-可见吸收光谱的实验值为275 nm, 计算值为269 nm, 误差为6 nm, 误差百分比小于3%[见图4(b)]。 图4(a)中的240~320 nm范围内的吸收峰为GHRP-6的本征吸收峰, 由分子结构中π → π * 轨道跃迁产生, GHRP-6分子收到光子激发后, 氨基酸残基中的离域π 键被激发跃迁至反键轨道, 产生279 nm的紫外吸收本征吸收峰; 催产素的紫外-可见吸收光谱中[图4(b)], 主要贡献为结构中酪氨酸上的π → π * 轨道跃迁。 在200~240 nm范围内存在的吸收谱带是由于电子跃迁过程中相互碰撞引起二次跃迁产生的, 不属于分子的本征吸收峰。 由此通过实验验证了该方法建立紫外-可见吸收光谱理论模型的准确性和可行性。
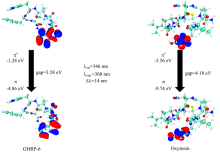
采用两种方法计算多肽类物质的荧光光谱: (1)根据分子前线轨道图中芳香族氨基酸的π * → π 轨道跃迁间的gap值, 预测分子的荧光峰位置; (2)对单重态第一激发态进行优化, 获得结构的第一激发态到基态的辐射能量, 根据高斯展宽得到多肽类物质的荧光光谱。
通过计算得到GHRP-6, Oxytocin的分子前线轨道能量, GHRP-6中位于色氨酸残基的π * → π 轨道跃迁为GHRP-6产生荧光的主要贡献; Oxytocin中位于酪氨酸残基的π * → π 轨道跃迁为Oxytocin产生荧光的主要贡献。 如图5(a)所示, GHRP-6中色氨酸残基上的π * → π 轨道间的gap值为3.58 eV, 计算得到产生荧光的波长为346 nm, 与实验值360 nm间误差为14 nm, 误差百分比约为4%; 如图5(b)所示Oxytocin中酪氨酸残基上的π * → π 轨道间的gap值为4.18 eV, 计算得到产生荧光的波长为296 nm, 与实验值312 nm间误差为16 nm, 误差百分比约为5%。 通过此方法, 可以模拟出分子中产生荧光的主要贡献位置, 在不考虑电子相关的情况下, 可以较为准确的预测出分子发出荧光所在的波长范围, 对分子荧光的理论计算和实验具有指导意义。
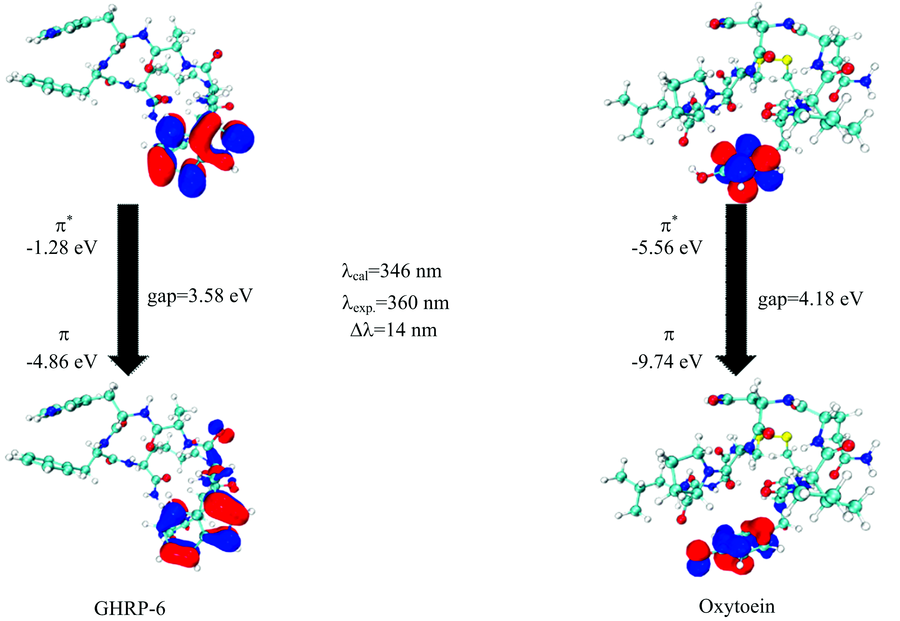 | 图5 GHRP-6(a)和Oxytocin(b)分子前线轨道的π * → π 跃迁Fig.5 The π * → π transition of the molecular frontier orbital of GHRP-6 (a) and Oxytocin (b) |
通过计算得到GHRP-6和Oxytocin从第一激发态跃迁到基态的跃迁能, 通过高斯展宽后得到的荧光光谱。 GHRP-6荧光光谱计算值为368 nm, 实验值为360 nm, 与实验值的误差为8 nm, 误差百分比小于3%; Oxytocin荧光光谱计算值为305 nm, 实验值为312 nm, 与实验值的误差为7 nm, 误差百分比小于3%。 GHRP-6产生荧光的发射波长与色氨酸产生的荧光波长近似, 说明GHRP-6产生荧光的主要贡献为色氨酸残基上的π * → π 轨道跃迁, 色氨酸残基与相连原子的二面角为7.08° , 2.54° , 分子的共平面程度较高, 呈现较强的刚性平面结构, 会增加荧光强度。 GHRP-6分子结构受到紫外吸收本征激发光的照射, 分子被激发后, 跃迁至单重第一激发态(S1)的π * 轨道, 当分子跃迁回基态(S0)的 π 轨道时, 即发出360 nm波长的荧光。 Oxytocin荧光峰位置与酪氨酸产生的荧光波长相似, Oxytocin产生荧光的主要贡献为酪氨酸残基上的π 轨道跃迁, 酪氨酸残基与其相连的原子的二面角为14.37° , 刚性平面程度相比于色氨酸较低, 荧光强度弱于色氨酸。
通过对GHRP-6的激发态计算, 证明可以通过本计算方法针对较大体系的多肽类物质, 计算其激发态下的紫外吸收光谱和荧光光谱, 分析其激发态下电子光谱的发生机理, 且与实验值吻合程度较高, 误差均在5%以内。 由此理论和实验结果验证了采用密度泛函理论计算建立荧光光谱理论模型的可行性。
 | 图6 GHRP-6(a)和Oxytocin(b)的荧光光谱的计算值和实验值Fig.6 Fluorescence spectra of GHRP-6 (a) and Oxytocin (b) Solid: Calculation results; Short Dash: Experimental results |
采用DFT与TDDFT方法, 在不影响计算精度的情况下引入TDA, RI等近似, 对较大体系的多肽类物质进行激发态计算, 得到紫外吸收光谱与荧光光谱的理论计算模型, 研究其结构性质与发光机理。 并与实验值对比得到如下结论:
(1)通过计算GHRP-6和Oxytocin的紫外-可见吸收光谱, 并与实验值进行比较, 实验测得的GHRP-6的紫外-可见吸收光谱最大吸收波长为279 nm, 计算得到的最大吸收波长为282 nm, 误差为3 nm, 误差百分比小于2%; Oxytocin紫外-可见吸收光谱的实验值为275 nm, 计算值为269 nm, 误差为6 nm, 误差百分比小于3%。 可以看出紫外-可见吸收光谱计算结果与实验值吻合程度较高, 该模型能准确计算得到多肽类物质等较大体系分子的紫外-可见吸收光谱。
(2)根据分子前线轨道理论, 探究多肽基态到激发态的电子轨道跃迁类型, GHRP-6产生荧光的发射波长与色氨酸产生的荧光波长相近, 说明GHRP-6产生荧光的主要贡献为色氨酸残基上的π → π * 轨道跃迁, Oxytocin荧光峰位置与酪氨酸产生的荧光波长相近, Oxytocin产生荧光的主要贡献为酪氨酸残基上的π → π * 轨道跃迁。 可通过该方法分析激发态下电子光谱的发光机理, 预测分子荧光发射的波长范围, 分析激发态光学性质。
(3)通过计算GHRP-6和Oxytocin的荧光光谱, 并与实验值进行比较, GHRP-6荧光光谱计算值为368 nm, 实验值为360 nm, 与实验值的误差为8 nm, 误差百分比小于3%; Oxytocin荧光光谱计算值为305 nm, 实验值为312 nm, 与实验值的误差为7 nm, 误差百分比小于3%。 通过实验验证了利用该方法建立荧光光谱模型的准确性和可行性。
利用密度泛函理论研究了较大体系的多肽类物质激发态下的紫外-可见吸收光谱和荧光光谱, 建立了理论计算模型, 并与实验对比, 验证了模型的可行性, 为较大体系的多肽类物质的光谱实验提供理论依据。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|