

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
固体物质的红外光谱衰减全反射与透射测试方法的比较研究
[杨珊 , 蔡秀琴, 张怡丰]
, 蔡秀琴, 张怡丰]
, 蔡秀琴, 张怡丰]
|
|


作者简介: 杨 珊, 女, 1981年生, 渭南师范学院化学与材料学院教授 e-mail: yss12041981@163.com
红外(IR)光谱是进行物质结构分析常用的工具, 广泛应用于各种固态、 液态、 气态材料的分析检测。 由于测试方法和制样过程直接影响IR测试结果的准确性, 研究了比较常用的衰减全反射(ATR)法与透射(TR)法在固体物质的IR光谱测试中的优劣。 选取3种常见高分子、 3种无机物以及3种有机小分子化合物作为研究对象, 分别用ATR法和TR法测试其IR光谱。 结合制样过程及谱图解析, 对谱图中异常现象进行了深入剖析。 TR法制样复杂、 干扰因素多: 样品与KBr混合研磨过程易吸收空气中水分, 对含N—H和O—H基团的样品分析会造成干扰; 样品太浓或太厚谱图会出现平头峰, 导致谱图无法解析; 样品颜色太深重时, 样片透光性差, 吸收峰强度整体减弱。 与TR法相比, ATR法无需制样, 省时省力且减少了吸水的可能性。 ATR法吸收峰的整体强度小于TR法, 因方法原理不同造成的。 尽管ATR法吸收峰的波数小于TR法几到几十cm-1, 但出峰位置在合理范围内, 定性分析不受影响。 由于ATR法测试时光进入样品的深度很浅(仅2~15 μm), 故谱图不会出现平头峰。 由于短波长的光不能穿透样品那么深, 故ATR法谱图中吸收峰的强度随波长的减小而减弱, 导致官能团区峰强度显著减小、 指纹区峰强度大大增强, 此问题可通过测试软件的“ATR校正”功能修正, 也可在定量分析中加以利用: 以测试的原始谱图对指纹区吸收峰定量, 以校正后谱图对官能团区吸收峰定量。 相比之下, ATR法不受样品颜色、 形状、 厚度限制, 测试简便、 快速、 准确、 无损、 样品可回收, 在高分子、 颜色深重以及易吸湿物质的IR测试上有明显优势。 故推荐广泛使用ATR法进行固体IR光谱检测。
Infrared (IR) spectroscopy is a common tool for material structure analysis, which is widely used in the detection of various solid, liquid and gaseous materials. Since the testing methods and sample preparation process directly affect the accuracy of IR spectra, the comparative study on the attenuated total reflection (ATR) and transmission (TR) methods used in measuring the IR spectroscopy of solid substances was carried out in this paper. Three kinds of common polymers, 3 kinds of inorganic substances, and 3 kinds of organic small-molecule compounds were selected as the research objects, and their IR spectra were measured by both ATR and TR methods, respectively. Various abnormal phenomena in the IR spectra were deeply analyzed combining sample preparation process and spectrum analysis. The TR method is complicated in the sample preparation process and has many interfering factors: the sample is easy to absorb moisture from the air during mixing and grinding process with KBr, which will disturb the analysis of samples having N—H and O—H groups; the spectrum will appear flat-headed peaks and can't be analyzed as the sample is too thick, and the absorption intensity of the whole spectrum will decrease as the sample is dark colored. Compared with the TR method, the ATR method requires no sample preparation, which saves both time and labor and reduces the possibility of water absorption. Because the method principle is different, the overall absorption intensity of the ATR method is less than that of the TR method. Although the peak wavenumber of the ATR method is generally smaller than that of the TR method among several to dozens of cm-1, the peak position is in a reasonable range, and the qualitative analysis is unaffected. Because the depth of light enters the sample is limited in the ATR method, only 2~15 μm, no flat-headed peaks appear in the ATR-IR spectrum. Due to the short wavelength light cannot penetrate the sample too deep, thus, the absorption intensity in ATR-IR spectrum will weaken along with the decrease of wavelength, which leads to the peak intensity in functional group region decrease and in fingerprint region greatly increases, while this problem can be revised by the “ATR correction” function of software, and also can be used in the quantitative analysis: the original spectra for analyzing the peaks in the fingerprint region, and the revised spectra for analyzing the peaks in functional group region. In comparison, the ATR method is not limited by the color, shape and thickness of the sample, i.e., the measurement is easier, fast, accurate, non-destructive, and recyclable. It has obvious advantages in the IR spectra detection of polymer, dark colored and hygroscopic substances. Thus, it is recommended to widely use the ATR method for detecting IR spectra of solid substances.
红外(IR)光谱是进行物质结构分析的有力工具, 广泛适用于固态、 液态或气态样品的研究, 测试温度范围很宽[1, 2, 3]。 IR光谱测试具有用样量少、 不破坏试样、 快速、 简便、 重复性好、 准确度高等特点, 被广泛应用于食品、 药品、 化妆品、 文物、 宝石、 织物、 纸张、 涂料、 土壤、 煤、 石油、 生物科学和现代医学诊断等领域中各种材料的分析检测[1, 4, 5]。 然而, 不同领域的研究人员对于IR光谱测试方法的选择及其制样过程对测试结果的影响缺乏系统、 直观的了解, 导致测试费时费力或不能准确反映样品结构信息。
IR光谱法的测试技术包括透射(TR)、 衰减全反射(ATR)和漫反射(DRIFTS)[2]。 样品的性质决定了适宜的样品制备技术[2]。 IR光谱的常规测试方法是TR法, ATR法常用于测试某些透光很差的、 强吸收的或腐蚀盐窗的物质以及不溶、 不熔且粉碎困难的弹性物质、 表面涂层、 纸张、 纤维等, 而DRIFTS法应用很少[2, 3]。 通常, 固体样品IR测试的经典方法是采用KBr压片法制样、 TR法测试, 然而, 该方法存在以下不足: 制样中易引入水干扰, 压片繁琐且易薄厚不均, 样品不易回收, 某些样品在与KBr粉末研磨时会发生吸湿、 离子交换、 置换、 络合反应等固相反应而引起谱带变化[3]。 而ATR法则是完全非破坏性技术, 快速、 无损样品, 测试任何种类的固体样品都不需要分离和制样处理, 对样品的大小、 形状没有特殊要求, 可用于太厚或透光太强的材料以及材料表面的测试, 还可检测含水或潮湿的样品, 对于表面和界面的微小变化很灵敏[1, 5]。
鉴于固体物质红外光谱法应用广泛, 研究中选取3种高分子、 3种无机物、 3种有机小分子共9种固体物质作为IR测试对象, 比较TR法和ATR法在测试过程和测试结果上的差异, 以期对固体IR测试的方法选择给出合理的、 指导性建议。
聚乙二醇1000(PEG-1000), 亚铁氰化钾[K4Fe(CN)6·3H2O], 过硫酸铵[(NH4)2S2O8], 葡萄糖酸钠, 甲基丙烯酸十八烷基酯(SMA), 十二烷基硫酸钠(SLS), 皆为分析纯; 溴化钾(KBr), 光谱纯; 碳化硅(SiC)纳米颗粒, 实验室合成; 聚对苯二甲酸乙二酯(PET), 市售蔬果乐SGL-300M1透明水果盒; 聚乙烯(PE), 市售妙洁PE保鲜膜。
傅里叶变换红外光谱仪(Tensor II, 德国Bruker), ZnSe单点衰减全反射附件(美国Pike); BM-13B压片模具, FW-5A手动压片机(博天胜达)。
TR法: 对于粉末状、 膏状固体, 干燥后取1~2 mg与KBr以1:100~1:200的比例混合、 研磨, 置于压片模具中, 在8~10 MPa压力下保持30~60 s, 压成透明样片, 测试透射光谱; 对于薄膜状、 薄片状固体则裁剪成小块测试。 测试条件: 先以KBr片扫描背景, 再测试样品, 波数4 000~400 cm-1、 分辨率4 cm-1、 扫描16次、 光阑6 mm。
ATR法: 无论粉末状、 膏状还是薄膜、 薄片状固体皆直接测试全反射光谱, 试样用量以充分盖住ATR附件的晶体表面为宜, 测试前后用无水乙醇清洁晶体表面, 以空气为背景, 波数4 000~600 cm-1、 分辨率4 cm-1、 扫描16次、 光阑6 mm, 谱图进行AB→TR转换。
2.1.1 PEG-1000
白色膏状固体PEG-1000 (HO—[CH2CH2O]n—H)的IR谱图见图1, 对TR法谱图[图1(a)]中各吸收峰归属如下[1, 6]: 3 388 cm-1为末端—OH的伸缩振动ν (OH), 2 885 cm-1为CH2的C—H对称伸缩振动ν s(CH2), 1 462 cm-1为CH2的C—H弯曲振动δ (CH2), 1 348 cm-1为C—O—H面内变形振动δ (C—O—H), 1 114和953 cm-1分别为C—O—C的伸缩振动ν (C—O—C)和面内变形振动δ (C—O—C), 839 cm-1峰为—CH2O—的C—H面内变形振动δ (CH2O)。 ATR法对应各峰波数见图1(b), 其日本SDBS谱库中的IR谱图(简称标谱)(KBr压片)[7]中对应各峰依次为: 3 434~3 424, 2 876, 1 456, 1 352, 1 106, 953和842 cm-1。 两种方法所得IR谱图中峰位置和峰形与标谱基本一致, 但ν (OH)峰的强度皆小于标准谱图, 推测可能与样品分子量标称值不准确所致; 同一官能团的吸收峰位置, ATR法小于TR法约1~9 cm-1; ATR法中ν (OH)峰的强度远小于TR法, 但δ (C—O—C)和δ (CH2O)两峰的强度却明显比TR法大, 这是由于ATR法测试的不足所致[8]——由于短波长的光不能穿透样品那么深, 故而2 800~4 000 cm-1的吸收强度受到影响而大大减弱, 而指纹区却增强。
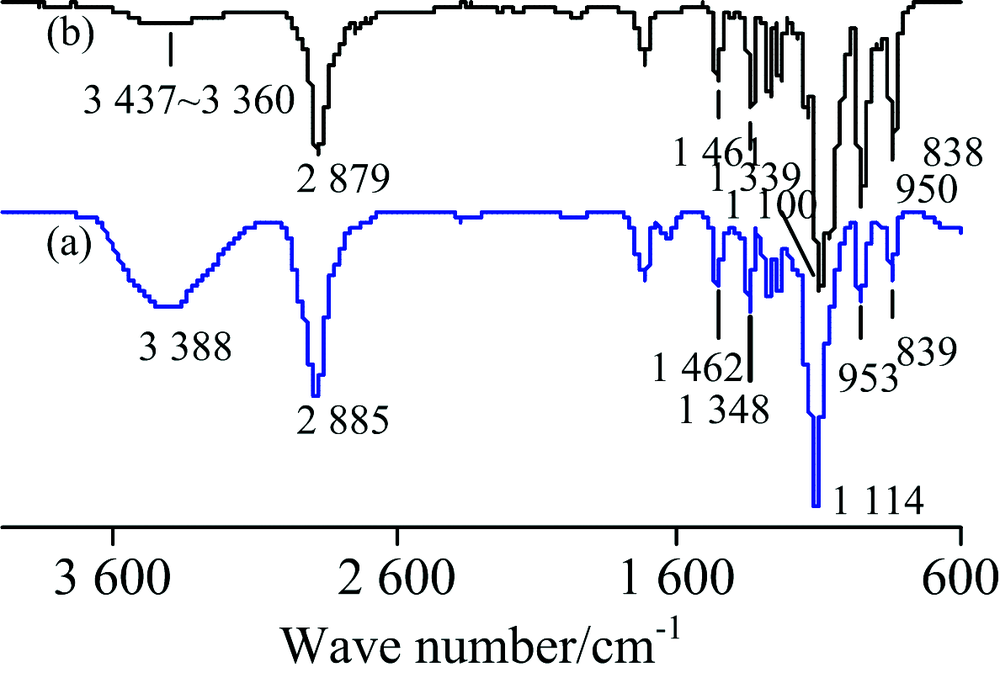 | 图1 PEG-1000的IR谱图 (a): TR; (b): ATRFig.1 IR spectra of PEG-1000 (a): TR; (b): ATR |
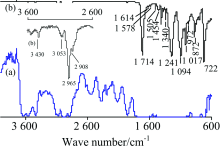
2.1.2 PET
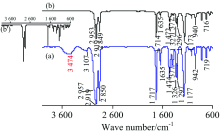
图2为PET (H—[OCH2CH2O—OC—C6H4—CO]n—OH)透明水果盒(厚0.32 mm)的IR谱图。 TR法谱图多处出现平头峰(即透光率T为0), 无法进行定性分析, 即使将PET的厚度降低至0.05 mm仍多处出现平头峰。 TR法中出现平头峰通常是由于样品浓度过大或样品太厚造成吸收太强所致, 而ATR法则由于光进入样品的深度很浅(仅2~15 μm)而不会出现平头峰[2]。 ATR法谱图与文献[8]一致, 对各峰归属如下[1, 9]: 3 430 cm-1极小峰为ν (O—H), 3 053 cm-1小峰为苯环上不饱和C—H的伸缩振动ν (=C—H), 2 965和2 908 cm-1为饱和CH2的ν as(CH2), 1 714 cm-1强峰为ν (C═O), 1 614, 1 578, 1 505和1 454 cm-1为苯环骨架C═C振动, 1 454 cm-1峰合并有CH2的弯曲振动δ (CH2), 1 340 cm-1为CH2的面外摇摆ω (CH2), 1 241和1 094 cm-1为ν as(C—O—C)和ν s(C—O—C), 1 017 cm-1为对位取代苯环C—H面内弯曲振动δ (=C—H); 972 cm-1峰为CH2—O的ν (C—O), 872和722 cm-1为苯环C—H面外弯曲振动δ (=C—H)。 ATR法中各峰位置与标谱(KBr压片)[7]吻合较好, 但高波数区的峰强度明显小于标准谱。
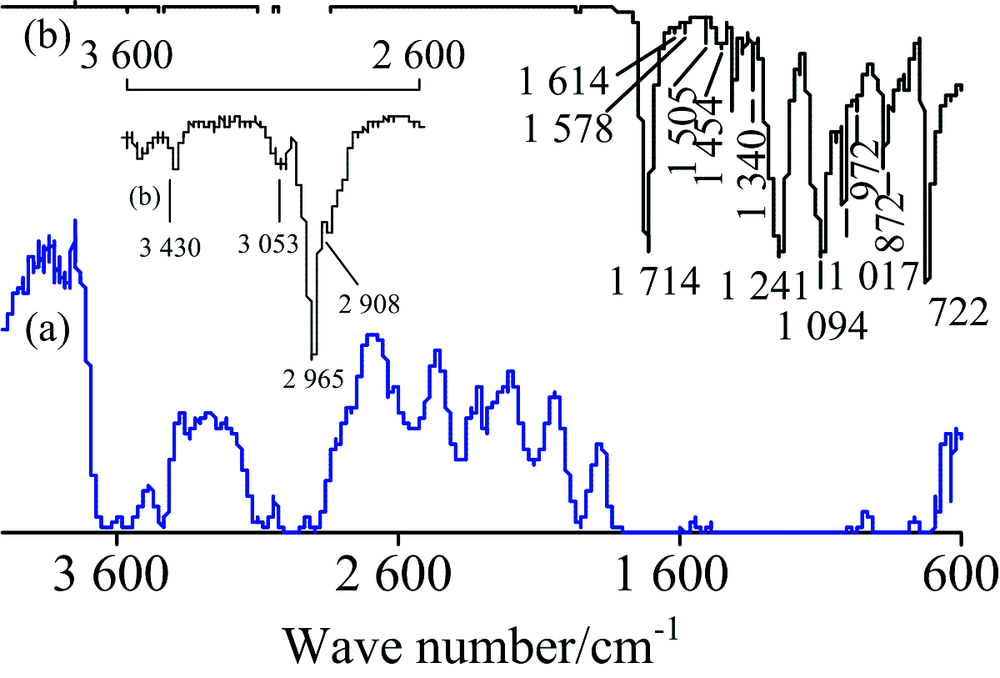 | 图2 PET的IR谱图 (a): TR; (b): ATRFig.2 IR spectra of PET (a): TR; (b): ATR |
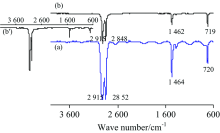
2.1.3 PE
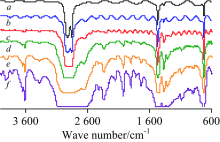
PE(—[CH2CH2]n—)薄膜(厚0.01 mm)的IR谱图见图3。 对TR法谱图[图3(a)]中各峰归属如下[1]: 2 915和2 852 cm-1分别为ν as(CH2)和ν s(CH2), 1 464 cm-1中强峰为CH2的弯曲振动δ s(CH2), 720 cm-1弱峰为—(CH2)n—(n≥ 4)的C—H面内摇摆振动ρ (CH2)n。 ATR法中各峰波数见图3b。 该PE膜的IR谱图与文献[8]报道一致, 表明市售保鲜膜为PE纯品。 对比可知: (1) TR法的基线很不平整, 由于膜太薄、 太软在膜夹具中很难像镜面一样平整, 因此存在光散射现象; (2) 同一官能团的吸收峰位置, ATR法波数小于TR法约3~7 cm-1; (3) ATR法中各官能团出峰的相对强度与TR法明显不同, 这与ATR法波长对吸收强度的影响有关[8], 该现象可通过IR测试软件的“ ATR校正” 功能消除[见图3(b')]; (4) TR法中ν as(CH2)峰为平头峰。 测试不同厚度PE膜的IR光谱, ATR法谱图完全相同, 而TR法谱图中平头峰的数目则随着厚度增大而增多(图4)。 事实上, 可溶或可熔的高分子膜或板可通过溶于适当溶剂或熔融的方法制备更薄的膜, 或者通过拉伸降低薄膜的厚度, 再用TR法测试; 但对于不溶不熔的高分子材料, 则只能用ATR法测试。
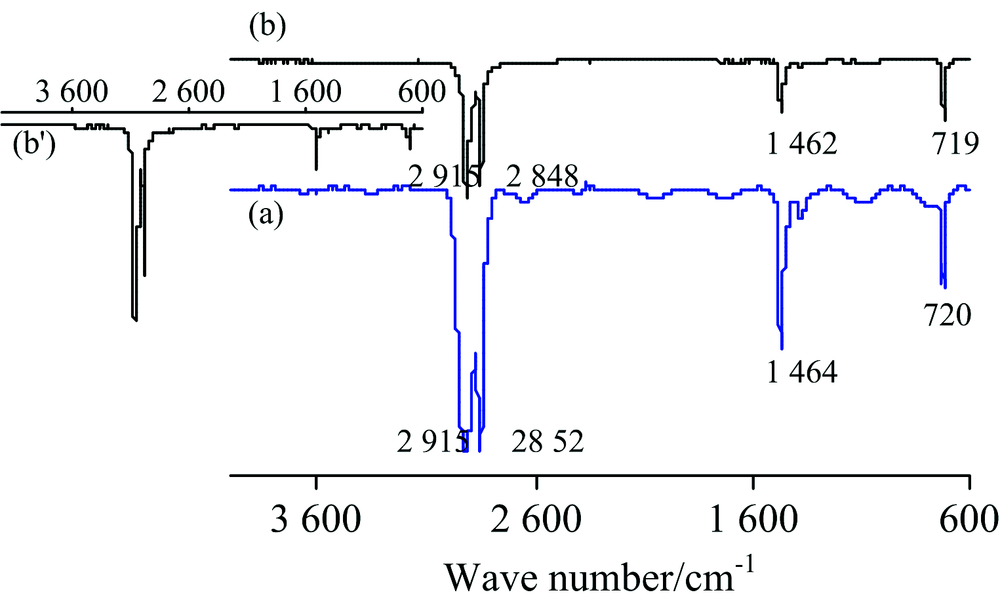 | 图3 PE的IR谱图 (a): TR; (b): ATRFig.3 IR spectra of PE (a): TR; (b): ATR |
 | 图4 不同厚度PE膜的TR-IR谱图 a: 0.01; b: 0.02; c: 0.04; d: 0.14; e: 0.37; f: 1.01 mmFig.4 TR-IR spectra of PE films with different thickness a: 0.01; b: 0.02; c: 0.04; d: 0.14; e: 0.37; f: 1.01 mm |
无机化合物的IR谱图比有机化合物要简单得多, 其在中红外区的吸收主要是由阴离子的晶格振动引起, 与阳离子的关系不大, 因此常常只出现少数几个宽吸收峰[1]。
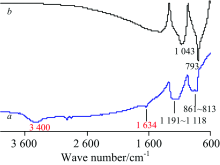
2.2.1 SiC
图5为SiC纳米颗粒(墨绿色)的IR谱, 对TR法谱(图5a)中各峰归属如下[1, 11, 12]: 3 400 cm-1包峰为水的ν (O—H), 1 634 cm-1为水的H—O—H弯曲振动δ (H—O—H), 1 191~1 118 cm-1为ν (Si—O), 861~813 cm-1为ν (Si—C); 而ATR法(图5b)中ν (Si—O)和ν (Si—C)分别在1 043和793 cm-1处, 未见水峰。 由于纳米级SiC在制备过程中表面易氧化, 存在少量SiO2, 因而谱图中出现ν (Si—O)峰[11]。 其标谱(石蜡糊法)[7]在1 090~1 085, 834~828 cm-1出峰。 TR法中ν (Si—O)和ν (Si—C)的波数皆大于标谱, 而ATR法则小于标谱, 二者的差异较大(几十cm-1), 但均在合理范围内[1]; TR法的基线不平、 吸收弱, 这是由于样品颜色深、 透光差; 两种方法的峰形和峰强差异较大, ATR法中ν (Si—C)峰尖且强, 与ATR法波长对吸收强度的影响有关[8]; TR法中H2O峰源于样品研磨过程中KBr吸水。
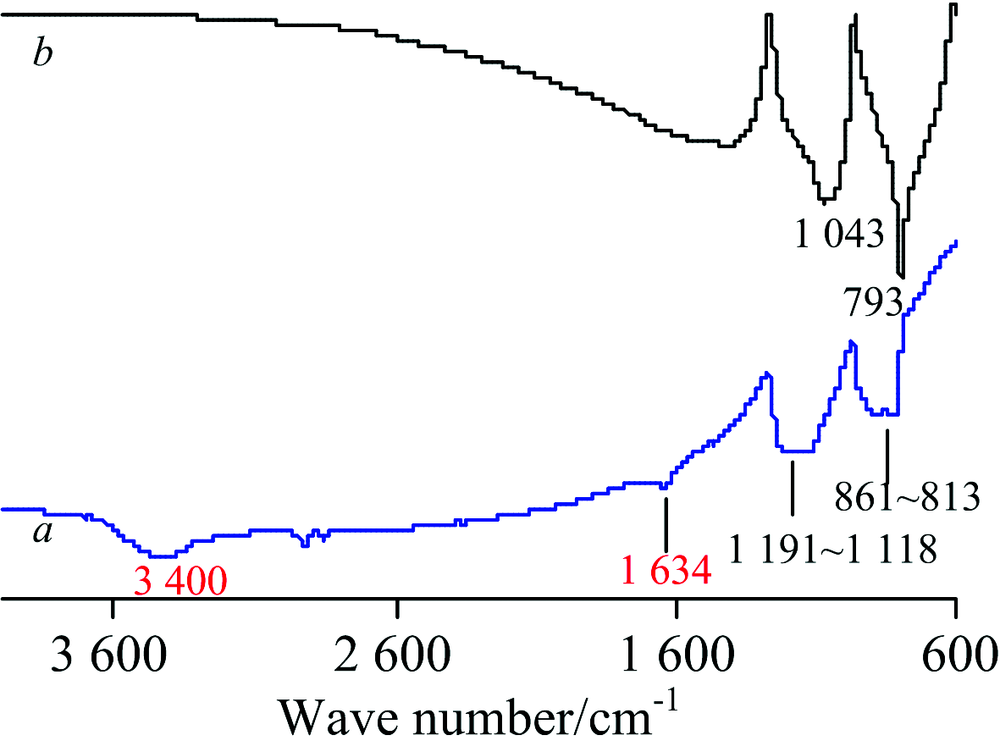 | 图5 SiC的IR谱图 a: TR; b: ATRFig.5 IR spectra of SiC a: TR; b: ATR |
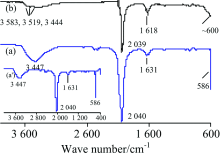
2.2.2 K4Fe(CN)6·3H2O
图6为K4Fe(CN)6·3H2O(浅黄色)的IR谱图, 其标谱(KBr压片)[7]在3 682, 3 524, 3 451, 2 043, 1 619和587 cm-1处出峰。 对TR法各峰归属[10, 12]如下: 3 447 cm-1包峰为水的ν (O—H), 2 040 cm-1强峰为ν (C═N), 1 631 cm-1为水的δ (H—O—H), 586 cm-1为ν (Fe—C)[见图6(a')]; 而ATR法[图6(b)]中3 583, 3 519和3 444 cm-1为结晶水的ν (O~H), 2 039 cm-1为ν (C═N), 1 618 cm-1为结晶水的δ (H—O—H), ~600 cm-1处未完全出峰为ν (Fe—C)。 尽管ATR法中结晶水的ν (O—H)峰比标谱小得多, 但整体上ATR法的峰形和峰位置与标谱符合较好, 而TR法受制样水干扰, 结晶水的出峰细节被掩盖。
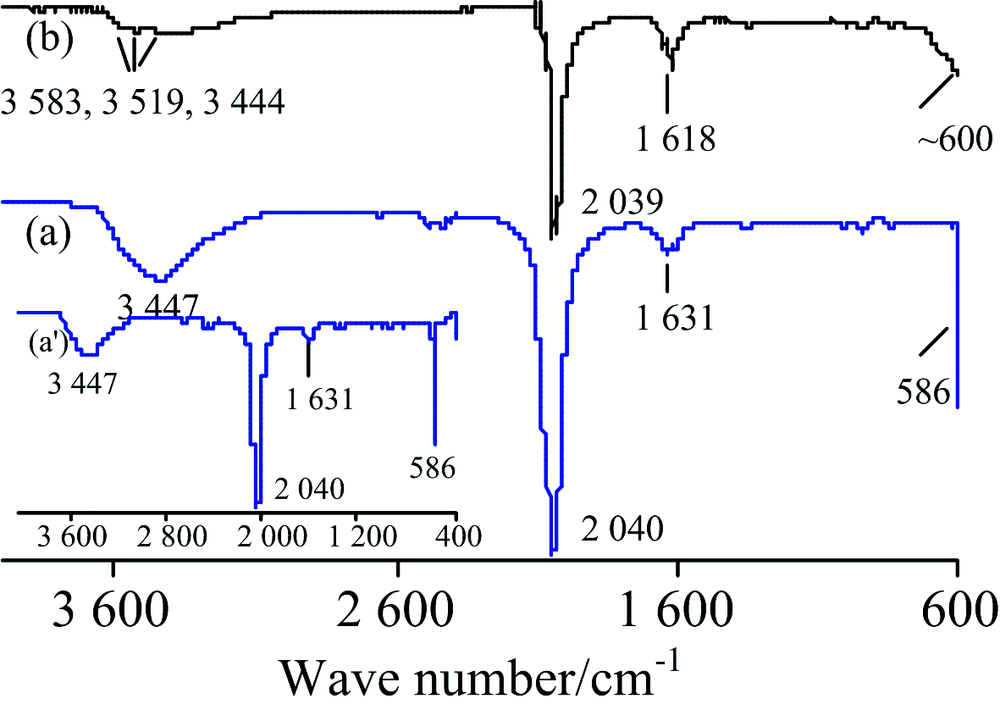 | 图6 K4Fe(CN)6·3H2O的IR谱图 (a): TR; (b): ATRFig.6 IR spectra of K4Fe(CN)6·3H2O (a): TR; (b): ATR |
2.2.3 (NH4)2S2O8
图7为(NH4)2S2O8(白色)的IR谱图。 据文献[10]报道, (NH4)2S2O8的IR谱图是由N
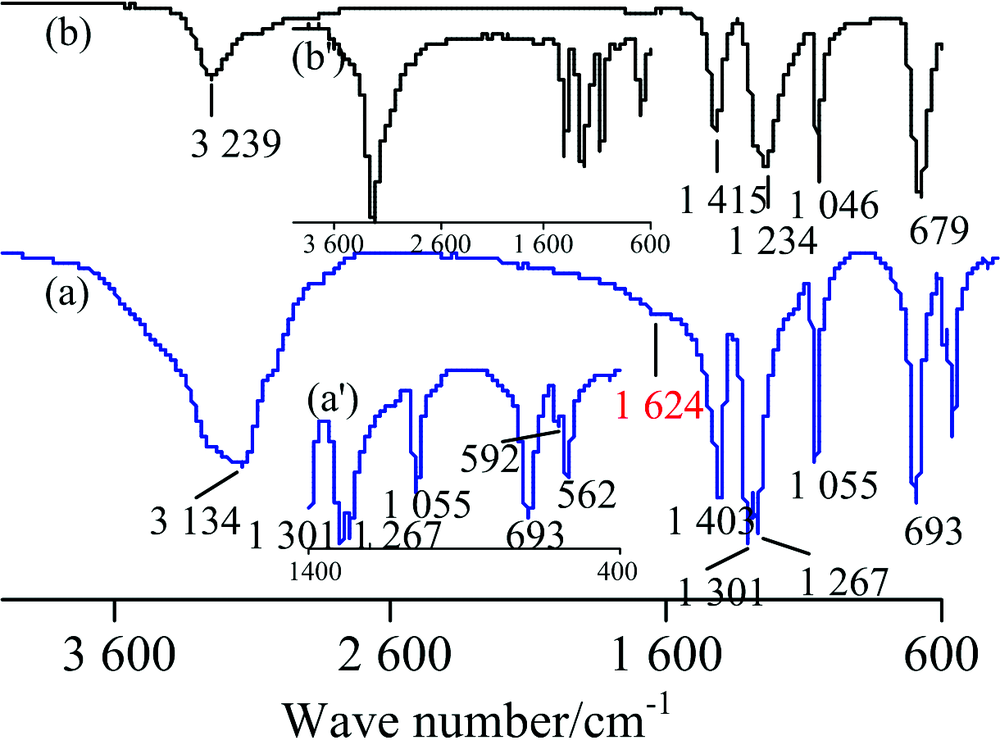 | 图7 (NH4)2S2O8的IR谱图 (a): TR; (b): ATRFig.7 IR spectra of (NH4)2S2O8 (a): TR; (b): ATR |
2.3.1 葡萄糖酸钠
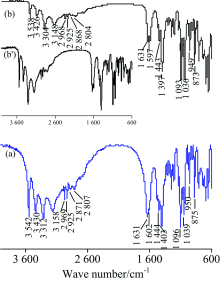
图8为葡萄糖酸钠(五羟基己酸钠, HOCH2(CHOH)4COONa)的IR谱图。 对TR法[图8(a)]中各峰归属如下[1]: 3 542, 3 430, 3 312和3 158 cm-1为ν (O—H), 2 969和2 925 cm-1为ν as(C—H), 2 871和2 807 cm-1为ν s(C—H), 1 631和1 602 cm-1为ν as(COO-), 1 474, 1 443和1 397 cm-1为ν s(COO-), 其中1 443 cm-1还合并C—H面内弯曲振动δ (C—H), 1 096和1 039 cm-1为ν (C—O)和ν (C—C), 949和873 cm-1峰为O—H的弯曲振动。 ATR法各吸峰波数见图8(b), 与TR法仅几个cm-1的差异。 两种方法的谱图皆与标谱[8]吻合较好; ATR法在官能团区的峰强比TR法小得多, 但经“ ATR校正” 后高频区出峰细节更清楚[见图8(b')]。 由于ATR法的出峰强度和样品与晶体接触的紧密程度有关[8], 为保证高波数区充分出峰且峰较强, 在测试时应确保充足的压力让样品与晶体接触良好。
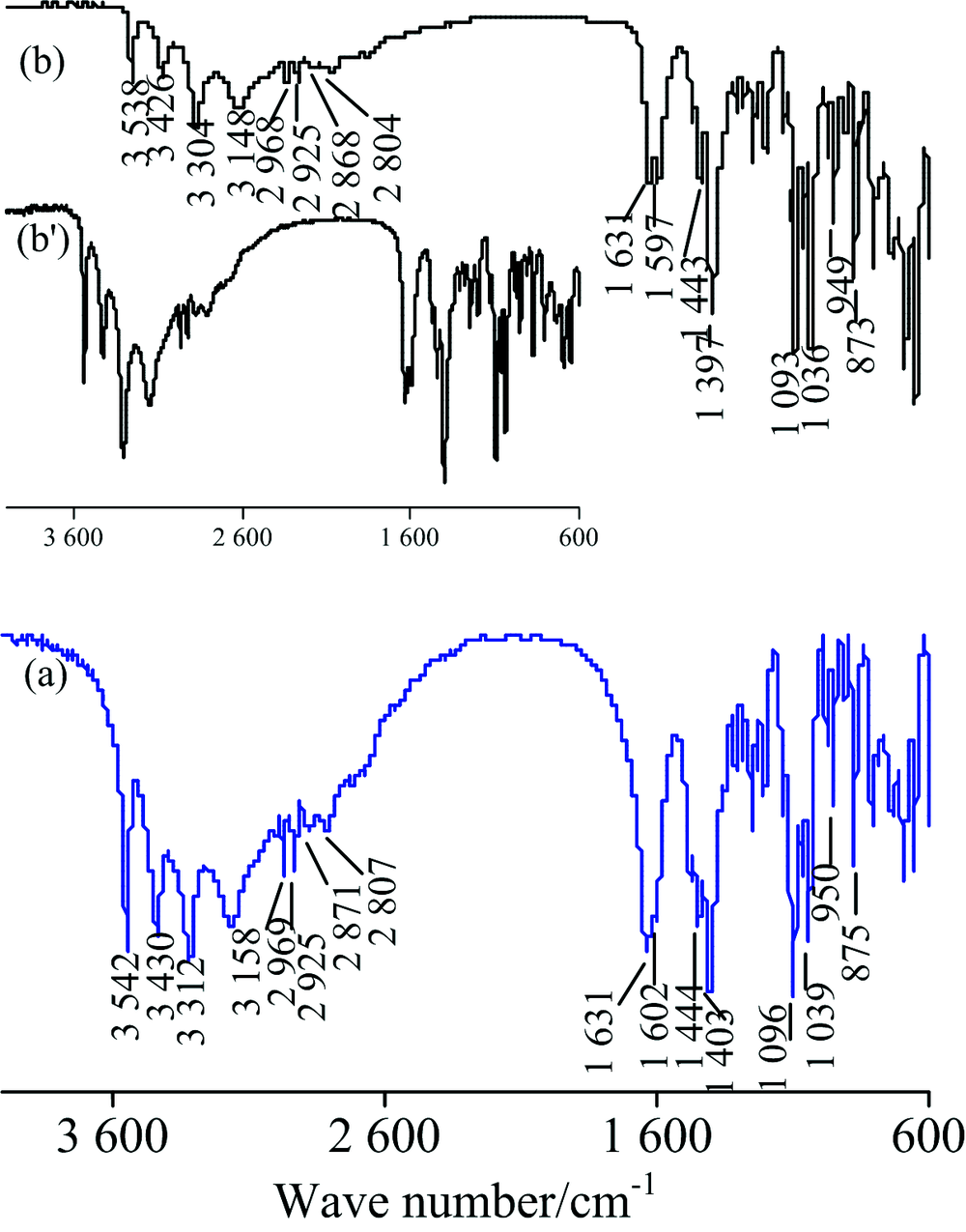 | 图8 葡萄糖酸钠的IR谱图 (a): TR; (b): ATRFig.8 IR spectra of sodium gluconate (a): TR; (b): ATR |
2.3.2 SMA
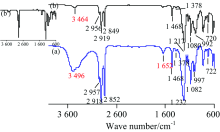
图9为SMA (CH2=C(CH3)COO(CH2)17CH3)的IR谱图, 对TR法[图9(a)]各峰归属如下[1]: 3 474 cm-1为水的ν (O—H), 3 107 cm-1为ν as(=CH2), 2 957 cm-1为ν as(CH3), 2 919 cm-1第一强峰为ν as(CH2), 2 850 cm-1为ν s(CH2), 1 717 cm-1为ν (C═O), 1 635 cm-1为ν (C═C), 1 474 cm-1为CH2的剪切振动δ (CH2), 1 377 cm-1小峰为—CH3的C—H对称变形振动δ s(CH3), 1 326和1 297 cm-1为=CH2的C—H变形振动δ (=CH2), 1 177 cm-1为ν as(C—O—C), 942 cm-1为=CH2的C—H面外弯曲振动τ(C—H), 719 cm-1为ρ (CH2)n。 ATR法未检出水峰和ν as(=CH2)峰, 其余各峰波数见图9(b), 由于端基烯ν as(=CH2)太弱而未被ATR法检出, 但可由1 635 cm-1处的ν (C═C)峰和1 326, 1 296和938 cm-1处δ (=CH2)峰确认双键的存在。 两种方法谱图的峰位置皆与标谱[7]符合较好。 与标谱[7]相比, ATR法中高波数区的峰强较小, 而指纹区的峰强较大, 通过“ ATR校正” 后[见图9(b')]峰强与标谱一致。 由于SMA疏水性强, 故TR法中水峰源于制样过程中KBr吸水所致, 吸水较多时水峰会将ν as(=CH2)掩盖。
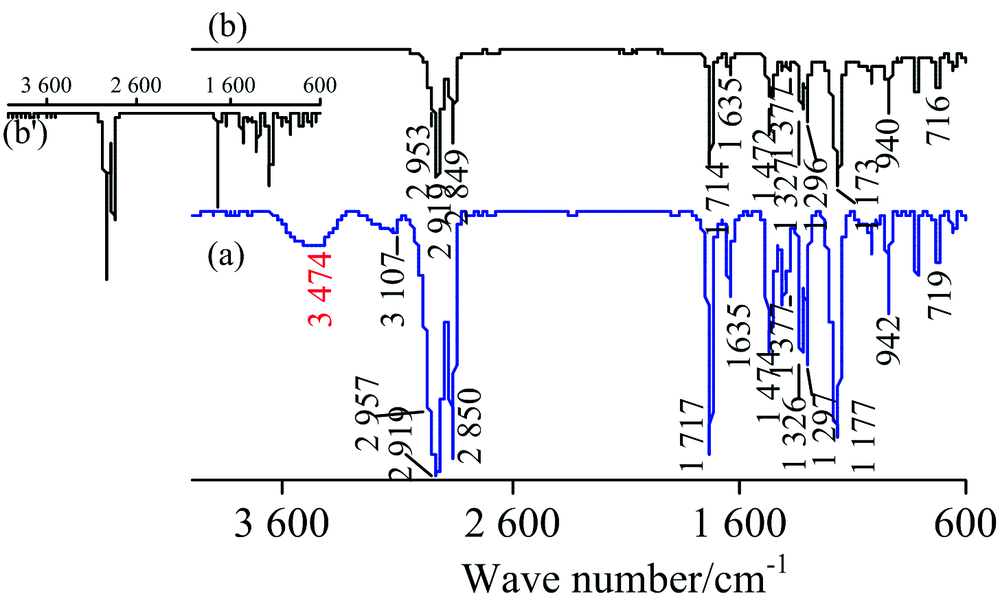 | 图9 SMA的IR谱图 (a): TR; (b): ATRFig.9 IR spectra of SMA (a): TR; (b): ATR |
2.3.3 SLS
图10为SLS [CH3(CH2)11SO4Na]的IR谱图, 对TR法[图10(a)]各峰归属如下[1, 13, 14]: 2 957 cm-1为ν as(CH3), 2 918 cm-1为ν as(CH2), 2 852 cm-1为ν s(CH2), 1 468 cm-1为CH2的剪切振动δ (CH2), 1 378 cm-1小峰为—CH3的对称变形振动δ s(CH3), 1 232 cm-1为ν as(S=O), 1 082 cm-1为ν s(S=O), 997 cm-1为ν (C—O—S), 722 cm-1为ρ (CH2)n。 由于SLS结构中有11个CH2, 故ν as(CH2)为第一强峰; 而ν s(CH3)[(2 870±10) cm-1]由于—CH3少、 峰太弱而被ν s(CH2)峰掩盖; 3 496和1 652 cm-1分别为水的ν (O—H)和δ (H—O—H)。 ATR法中3 464和1 658 cm-1为水峰, 其余各峰波数见图10(b)。 比较可知, 两种方法的谱图出峰位置皆与标谱[7]符合较好; ATR法高波数区的峰强相对较小, 经“ ATR校正” 后[图10(b')]与TR法一致; ATR法中水峰表明样品含水, 这是由于SLS作为阴离子表面活性剂有较强的吸水能力, 而TR法水峰很大表明样品在研磨中还有吸水。 对于易吸湿的样品, 在测试前必须进行干燥处理, 使用TR法制样时研磨及转移至模具过程尽量在红外灯照射下操作, 可大大减少样品或KBr吸水。
 | 图10 SLS的IR谱图 (a): TR; (b): ATRFig.10 IR spectra of SLS (a): TR; (b): ATR |
比较研究了TR法和ATR法在3种常见高分子、 3种无机物以及3种有机小分子化合物的固体IR测试中的差异。 TR法制样复杂、 易受水干扰, 样品太浓、 太厚谱图会出现平头峰, 样品颜色太深重则吸收大大减弱。 相比之下, ATR法无需制样, 不受样品颜色、 形状、 厚度限制, 测试简便、 快速、 准确、 无损, 样品可回收。 鉴于ATR法在固体IR测试中优势明显, 推荐广泛使用ATR法进行固体IR光谱检测。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|